Reactivity in Chemistry
Aliphatic Nucleophilic Substitution
NS20. Solutions to selected problems
Problem NS1.1.
Problem NS1.2.
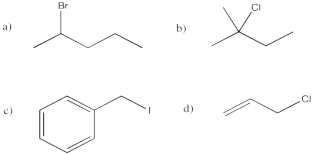
Problem NS1.3.
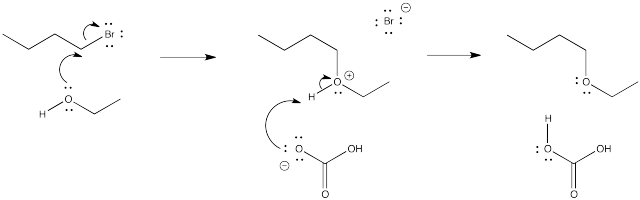
Problem NS1.4.
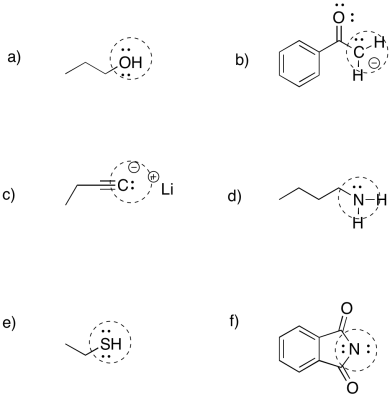
Problem NS2.1.
The electronegativity of carbon (2.55 on Pauling scale) is less than that of fluorine (3.98), chlorine (3.16), bromine (2.96) or iodine (2.66).
a) On that basis, the carbon attached to a halogen is electrophilic because it has a partial positive charge resulting from the polar carbon-halogen bond.
b) We would expect an alkyl fluoride to be the most electrophilic of these compounds, based on electronegativity.
c) Assuming the energy required for breaking the carbon-halogen bond plays a major role in the activation barrier (not guaranteed), we would expect the activation barrier to be lowest with the alkyl iodide, then the alkyl bromide, then the alkyl chloride and finally the alkyl fluoride. This prediction contrasts with what we might expect based on electronegativity.
d) The stability of alkyl fluorides towards this reactions suggests that there is, in fact, a prominent role played by bond strengths, at least in that case. The carbon-fluoride bond is strong enough to hinder nucleophilic substitution in this compound.
Problem NS2.2.
a) In mechanism B, the dissociative one, we would expect a higher activation enthalpy. The first step, which appears to be rate determining, is a bond-breaking step, which will cost energy. In mechanism C, the bond-breaking is compensated by some bond-making; overall, this probably costs less energy.
b) In mechanism B, the dissociative case, we expect a more positive entropy of activation. As the bond to the halide begins to break, the halide and carbocation fragments begin to move independently of each other, gaining degrees of freedom and increasing in entropy. In mechanism C, the incoming nucleophile appears to coordinate its motion with that of the departing halide; as a result, there are fewer degrees of freedom in this case.
Problem NS2.3.
a) Charged intermediates are present in the dissociative mechanism (B).
b) It seems like a more polar solvent would favour both mechanisms, because both involve the interaction of an anionic nucleophile with an electrophile and loss of an anionic leaving group. However, the dissociative case (B) involves a build-up of charge in the intermediate. It is possible that a more epolar solvent could reduce the barrier to that buildup of charge separation, accelerating this mechanism.
Problem NS2.4.
a) The rate-determining step is probably the bond-breaking one (the first one).
b) Because the nucleophile has not yet participated at that point, Rate = k [R-X], if R-X = the alkyl halide.
c) There is only one step; it is the rate-determining step, by default.
d) Rate = k [R-X][Nu].
Problem NS3.1.
a) The rate-determining step in SN1 does not depend on the nucleophile concentration, so the graph shows a flat line.
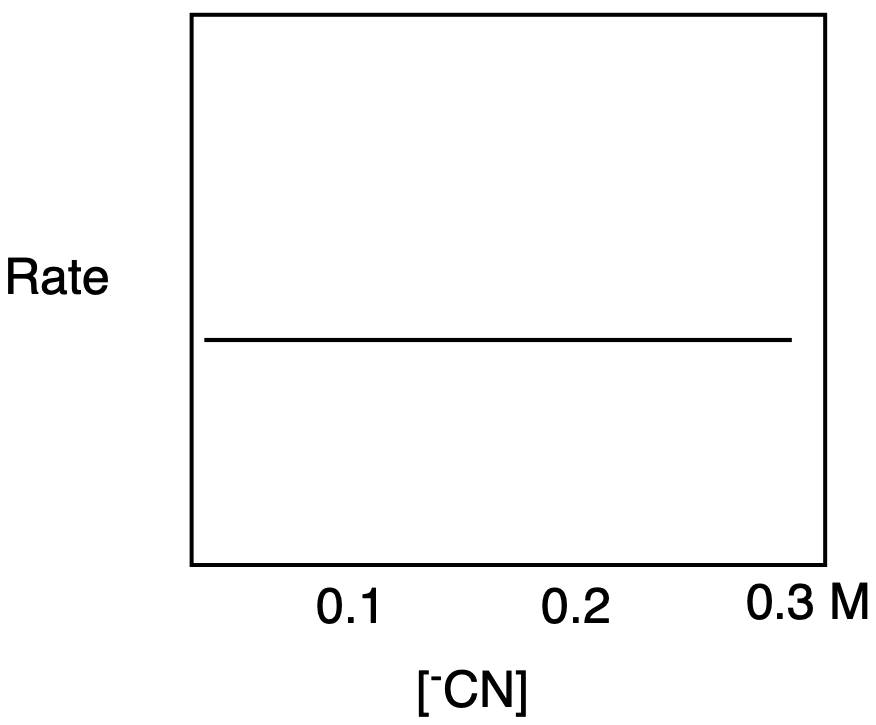
b) The rate-determining step in SN2 depends on the nucleophile concentration, so the graph shows a straight line with an upward slope.
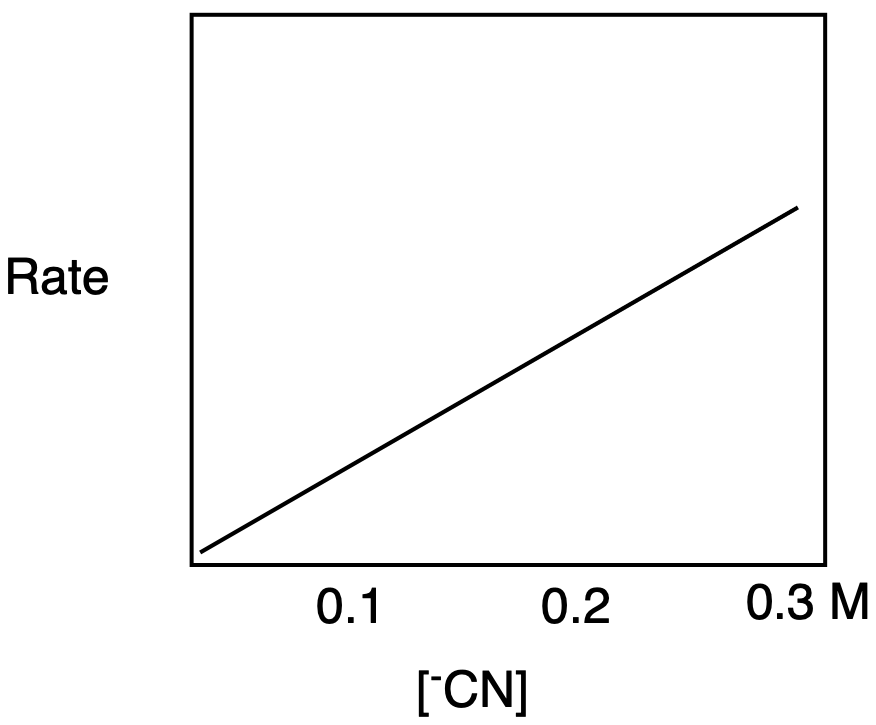
b) The rate-determining step in SN1 depends on the electrophile concentration, so the graph shows a straight line with an upward slope.
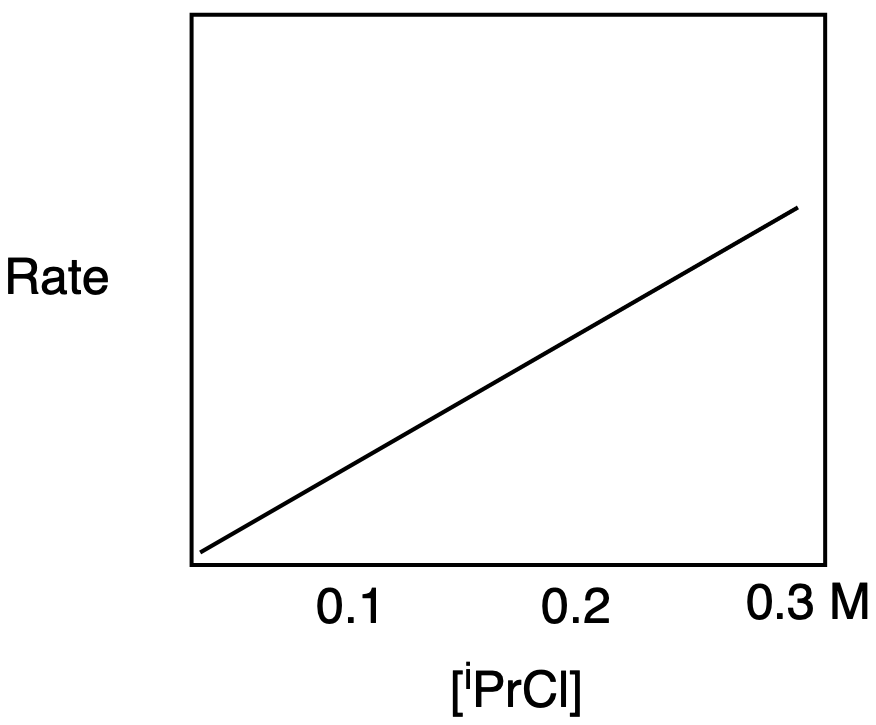
b) The rate-determining step in SN2 depends on the electrophile concentration, so the graph shows a straight line with an upward slope.
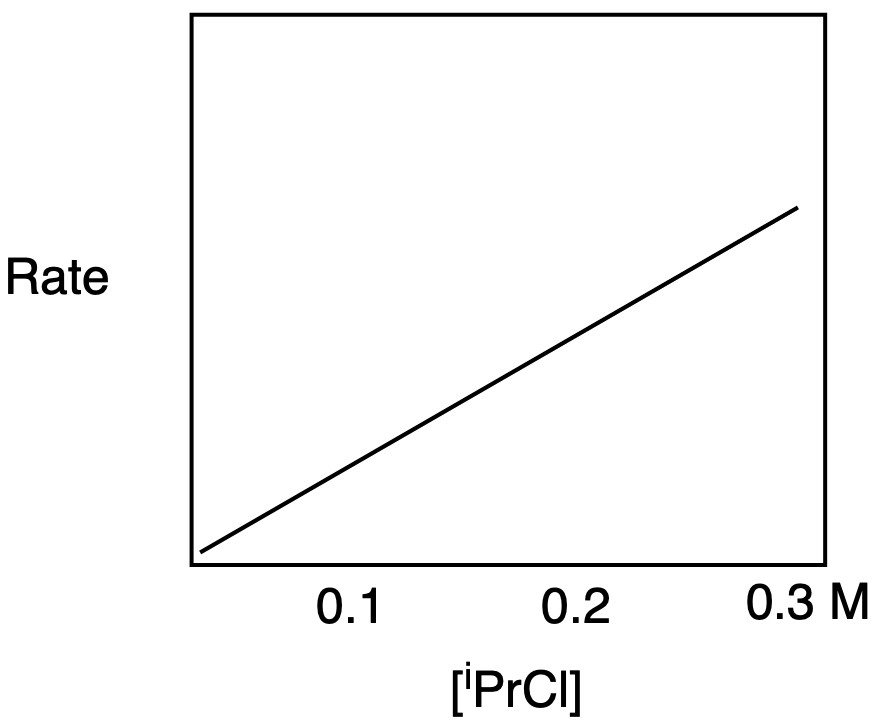
Problem NS3.2.
Steric effects or crowdedness at the electrophilic carbonplays a strong role in an SN2 reaction because the nucleophile must connect with this carbon leading to a transition state in which the carbon is 5-coordinate (surrounded by five different groups). Crowding at this carbon slows down the SN2 pathway.
Crowding is less important in an SN1 pathway because the carbon is trigonal planar when the nucleophile approaches, so there is more room for the incoming nucleophile.
Thus, steric crowding slows down the SN2 pathway but has little effect on the SN1 pathway.
Problem NS3.3.
There is a buildup of positive charge in the transitions state of the rate-determining step in an SN1 reaction because of the formation of a cationic intermediate. This positive charge is generally stabilized by hyperconjugative effects of surrounding alkyl groups. The more substituted the electrophilic carbon, the more stable the developing cation and the faster the reaction.
Similar cation-stabilizing effects also come about through resonance effects in the developing cation in an SN1 reaction. These charge-delocalizing effects are commonly seen in allylic cations (CH2=CHCH2+) and benzylic cations (PhCH2+).
There is often some buildup of negative charge in the transitions state in an SN2 reaction because of the approach of an anionic nucleophile, but this negative charge is generally stabilized by the electronegative leaving group.
Problem NS4.1.

Problem NS4.2.
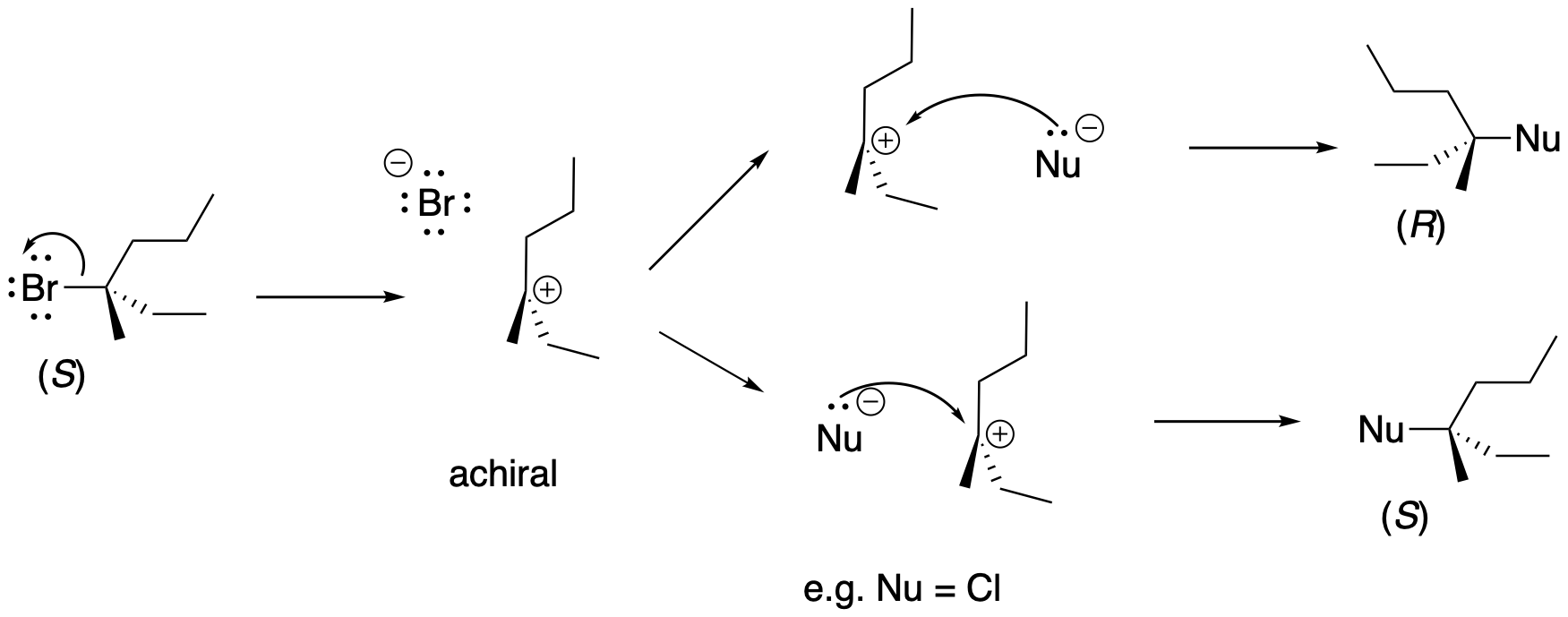
Problem NS4.3.
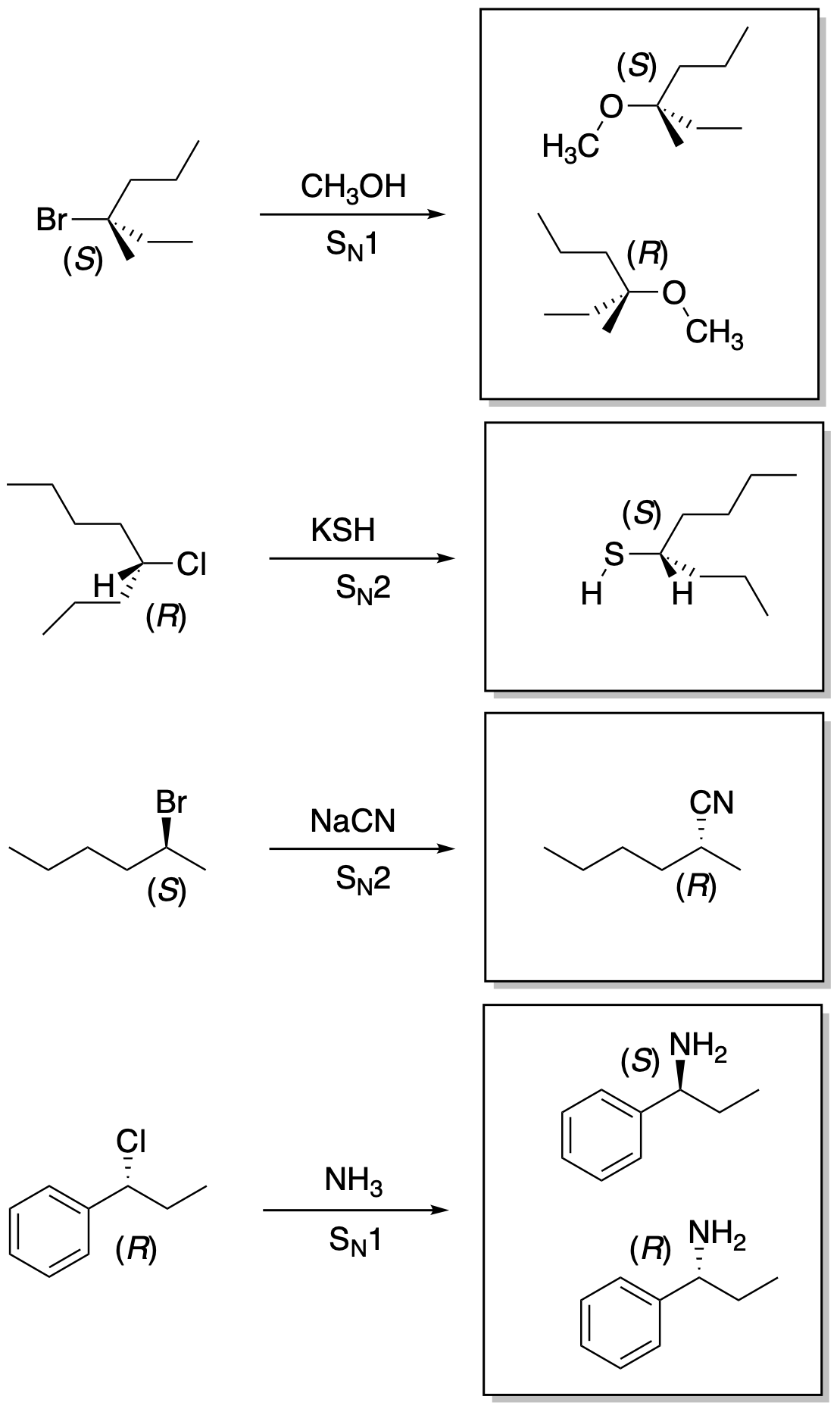
Problem NS4.4.
Most pharmaceuticals bind with proteins to either slow down or speed up a specific biochemical process in order to treat a medical condition. Proteins are vast molecules composed of chiral amino acid building blocks and the proteins are also chiral as a ressult. If a pharmaceutical is chiral, then one enantiomer fits into the binding site of the protein but the other enantiomer does not. When making a pharmaceutical, laboratories hope to make only the active enantiomer both for efficiency reasons (otherwise half of the material that they spend time and money making is worthless) and for medical reasons: limiting the drug to one enantiomer helps prevent any unexpected biological activity from the other enantiomer.
Problem NS5.1.
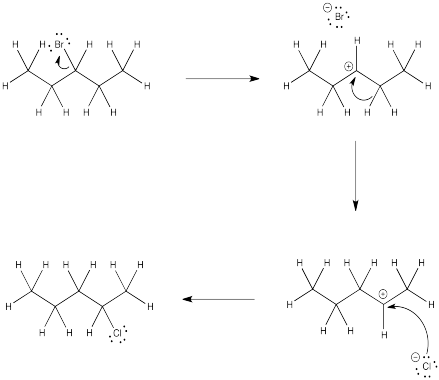
Problem NS5.2.
The initial cation formed upon ionization has the positive charge on carbon 3 of the chain. Rapid hydride shifts lead to other secondary cations at both carbon 2 and carbon 4. Although these two carbons are symmetry-equivalent, it is useful to think of them separately for statistical purposes. Combination of chloride ion with the cation at carbon 3 leads to 3-chloropentane. Commbination of chloride ion with the cation at either carbon 2 or carbon 4 leads to 2-chloropentane. There are twice as many cations of similar stability leading to 2-chloropentane compared to 3-chloropentane, so the percent 2-chloropentane = (2/(2+1))x100% = 67%.
Problem NS5.3.
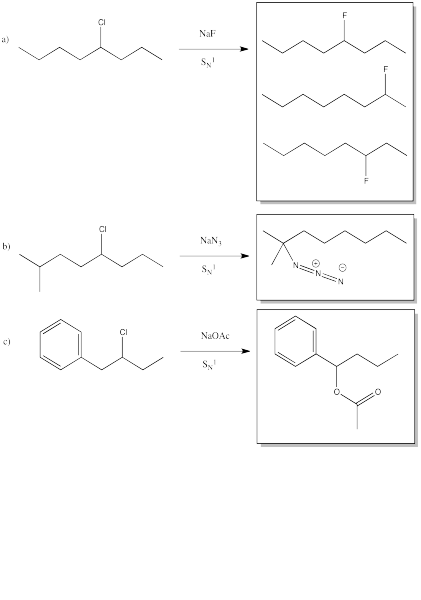
Problem NS6.1.
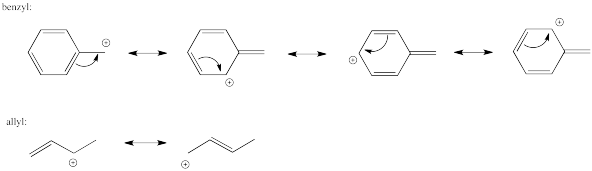
Problem NS6.2.
Keep in mind that there are other factors that can influence the reaction pathway; what we have here are just the most likely mechanisms.
a) SN2 b) Both pathways are very possible c) Both pathways are very possible d) SN2
e) SN2 f) SN2 g) SN1 h) SN1 i) SN1 j) SN1
Problem NS7.1.
a) ethanol, isopropanol, trifluoroacetic acid
b) hexane, toluene
c) THF, acetonitrile, DMF, dichloromethane, ether, DMSO, triethylamine, pyridine
d) DMSO > DMF > ACN > pyridine > DCM > THF > ether > TEA, based on dielectric constants. In general, the ones with multiple bonds between two different atoms are the most polar.
e) pyridine and triethylamine. The lone pair on the nitrogen atom is basic toward protons. The trend in basicity is triethylamine > pyridine >> acetonitrile; as the percent s character in the lone pair increases, the electrons are lower in energy and less available for donation.
Problem NS7.2.
a) Reaction of this benzylic alkyl halide in a polar, aprotic solvent should proceed at a medium speed via SN1.
b) Reaction of this tertiary alkyl halide in a protic solvent should be fast via SN1.
c) Reaction of this primary alkyl halide in a protic solvent should be slow via SN1.
Problem NS7.3.
a) Reaction of this secodary alkyl halide with a strong alkynide nucleophile in a polar, aprotic solvent should proceed at a medium speed via SN2.
b) Reaction of this tertiary alkyl halide with a strong azide nucleophile in a polar aprotic solvent should be very slow via SN2.
c) Reaction of this secondary alkyl halide with a phenolate nucleophile in a protic solvent should be slow via SN2.
Problem NS8.1.
a) The greater the charge on a nucleophile, the faster it reacts. For example, methoxide (CH3O-) is faster than methanol (CH3OH), and phenyl sulfide (PhS-) is faster than phenyl thiol (PhSH).
b) Comparing two atoms in the same column of the periodic table, we see that the greater the size of the atom bearing the lone pair, the faster it reacts. For example, phenyl sulfide (PhS-) is faster than phenoxide (PhO-), iodide (I-) is faster than bromide (Br-), and triphenylphosphine (Ph3P) is faster than triethylamine (Et3N).
However, remember that this trend reverses in aprotic or non-hydrogen bonding solvents. The other trends all remain the same.
c) Comparing two atoms in the same row of the periodic table, we see that the greater the electronegativity of the atom bearing the negative charge, the more slowly it reacts. For example, methanol (CH3OH) is slower than ammonia (NH3), and fluoride (F-) is slower than methoxide (CH3O-).
c) The greater the charge delocalization of the nucleophile, the more slowly it reacts. For example, phenoxide (PhO-) is slower than methoxide (CH3O-), and acetate (CH3CO2-) is slower than phenoxide (PhO-).
Problem NS8.2.
a) Nucleophilicity should play a stronger role in an SN2 reaction than in an SN1 reaction. In an SN1 reaction, the reaction rate is independent of the nucleophile. We normally see that lack of dependence in a rate law that neglects nucleophile concentration. The arrival of the nucleophile after the rate determining step of ionization means the reaction rate is already determined at that point, regardless of nucleophile structure.
b) Methyl bromide or bromomethane, CH3Br, is not likely to proceed through an SN1 substitution pathway because it does not yield a stable carbocation.
Problem NS8.3.
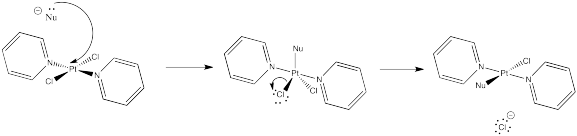
Problem NS8.4.
A very fast nucelophile may promote an SN2 pathway by acting on the electrophile before ionization has had a chance to occur. However, there are other factors, such as electrophile structure, that generally play a stonger role in determining the chance of SN2 vs. SN1 reaction.
Problem NS9.1.
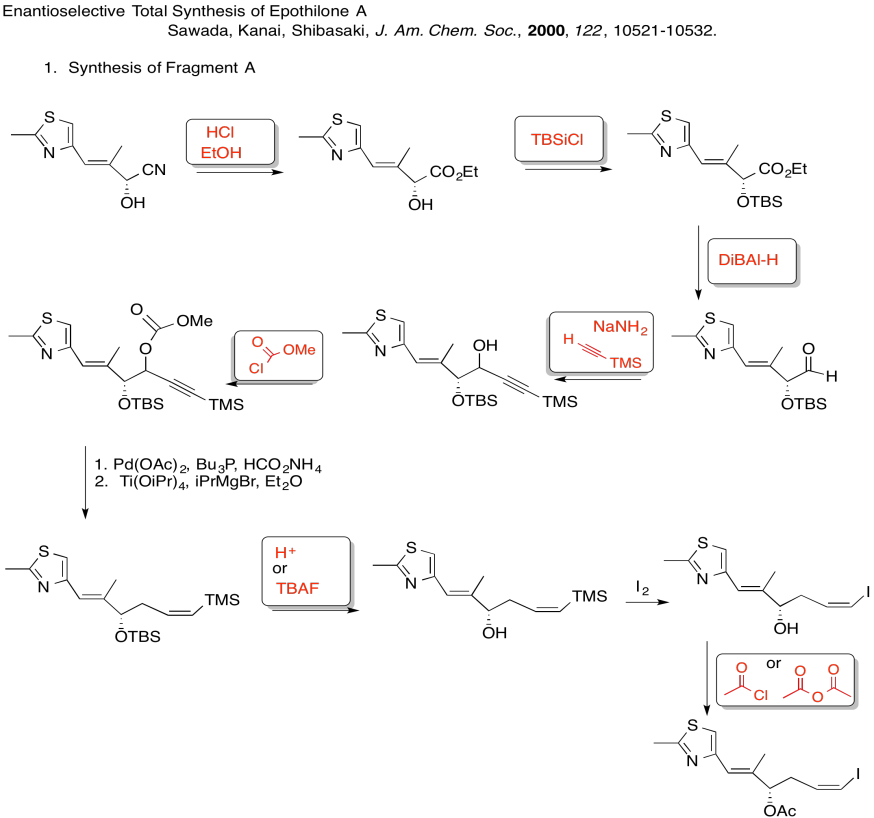
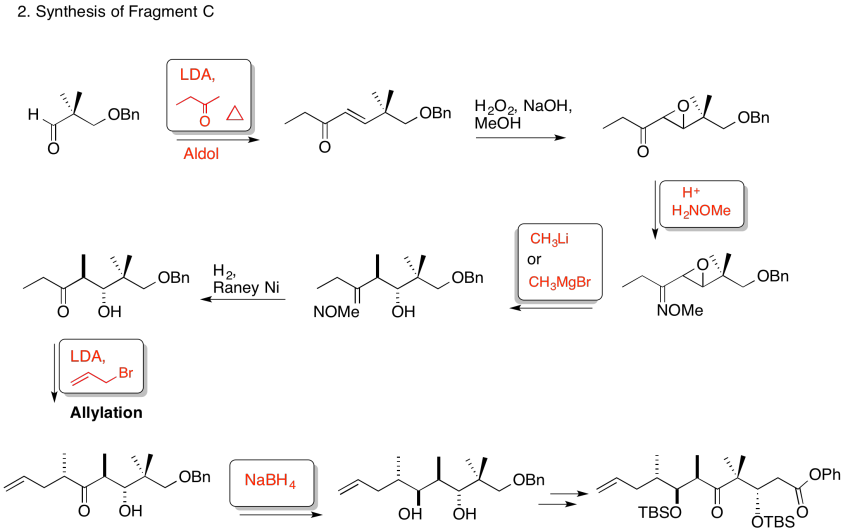
3. Kinetic -- to fully deprotonate (not equilibrate or you lose stereocontrol) and for the chelation control.
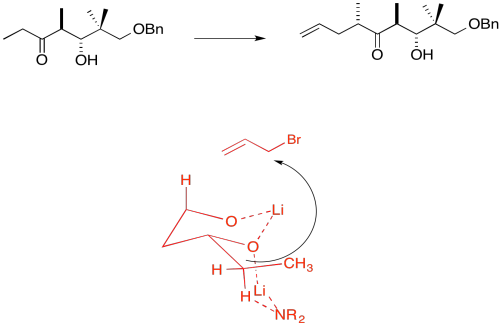
Due to chelation effects shown above, the LDA preferentially removes one hydrogen to form only the Z-enolate that will then do the SN2 to allyl bromide on only one face.

Problem NS10.1.
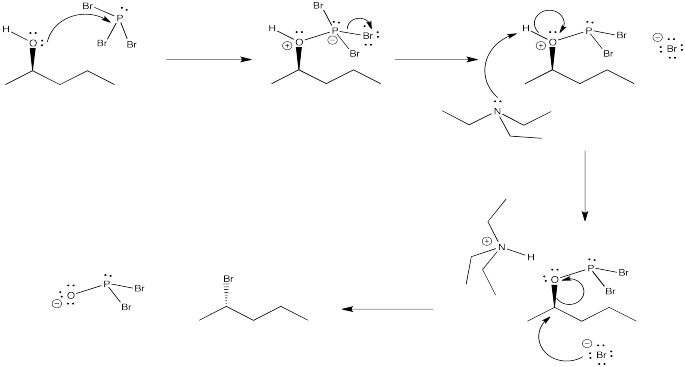
Problem NS10.2.
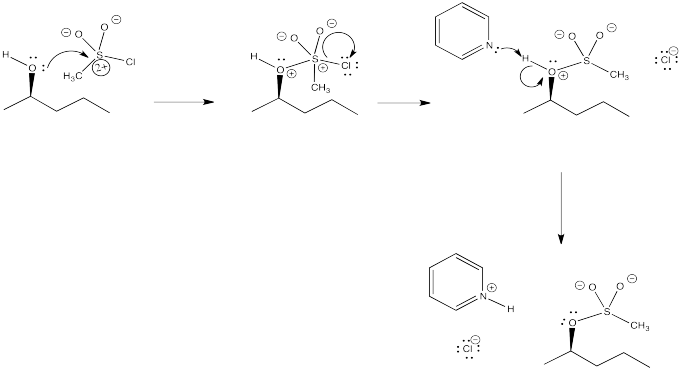
Problem NS10.3.
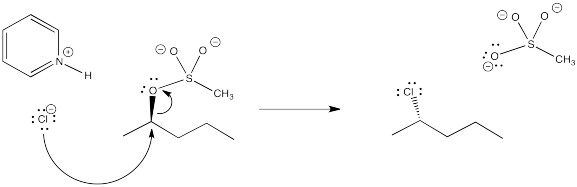
Problem NS10.4.
Problem NS10.5.
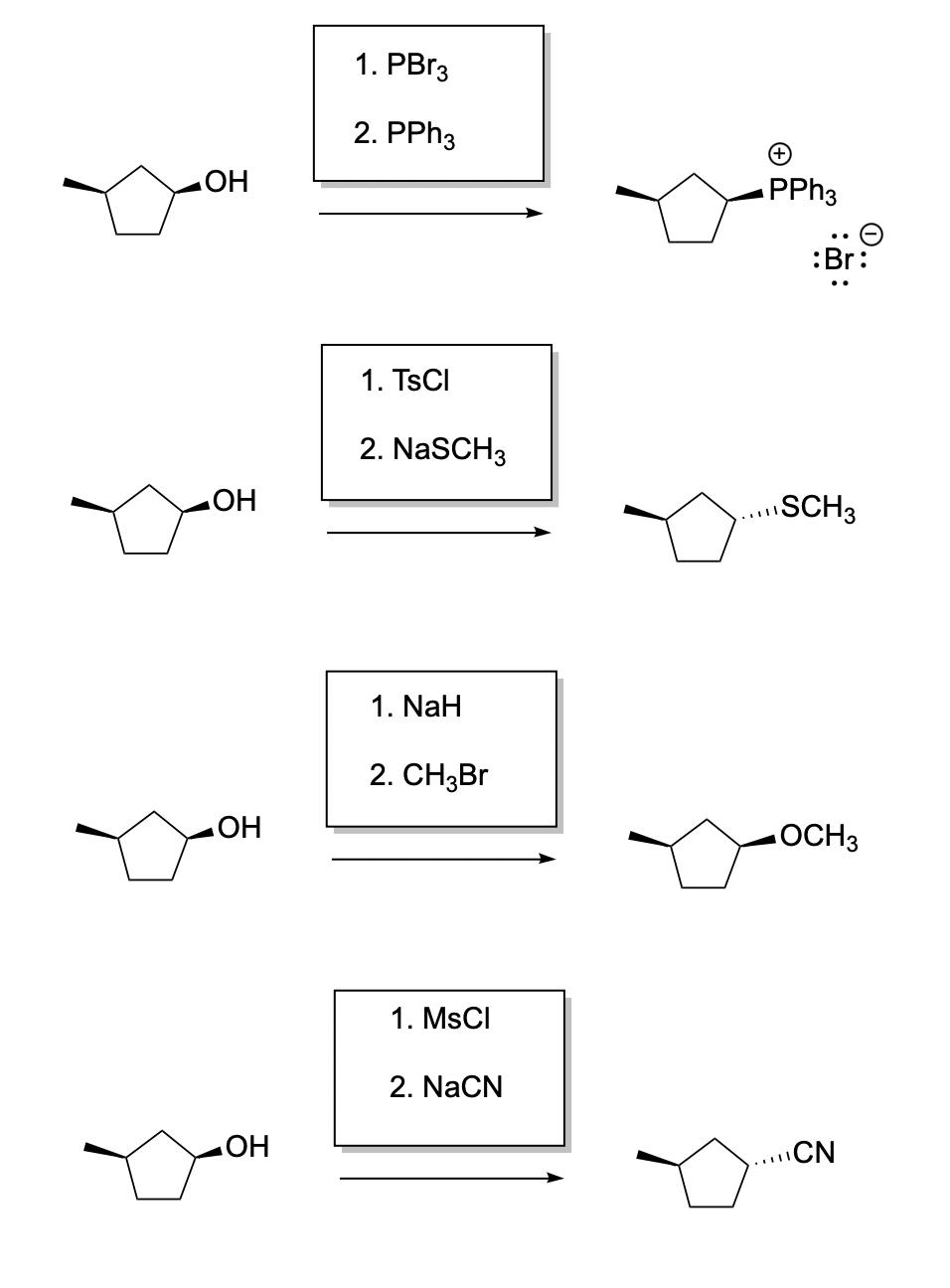
Problem NS10.6.
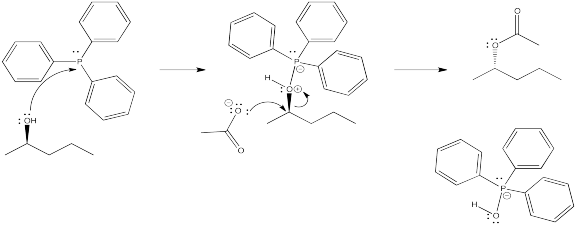
DEAD acts as an oxidizing agent to convert the phosphorus product to a stable side-product, triphenylphosphine oxide, Ph3P=O.
Problem NS11.1.
Problem NS11.2.
Problem NS11.3.
Problem NS11.4.
Problem NS12.1.
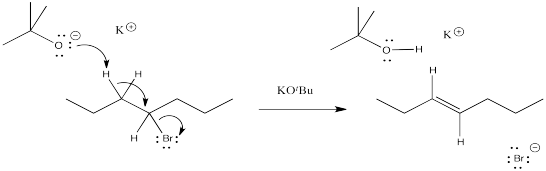
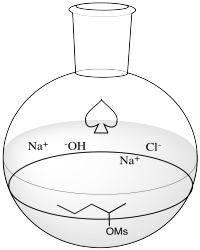
Problem NS12.2.
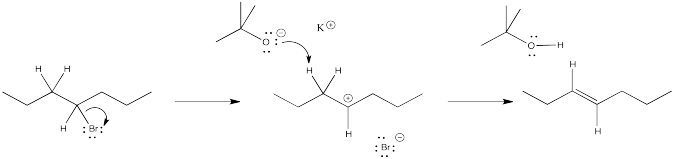
Problem NS12.3.
a) products of cation rearrangement via hydride shifts: 2-heptene instead of 3-heptene.
b) The absence of rearrangement suggests the absence of cations. The mechanism for the reaction shown must be concerted rather than via the ionic intermediate.
Problem NS12.4.
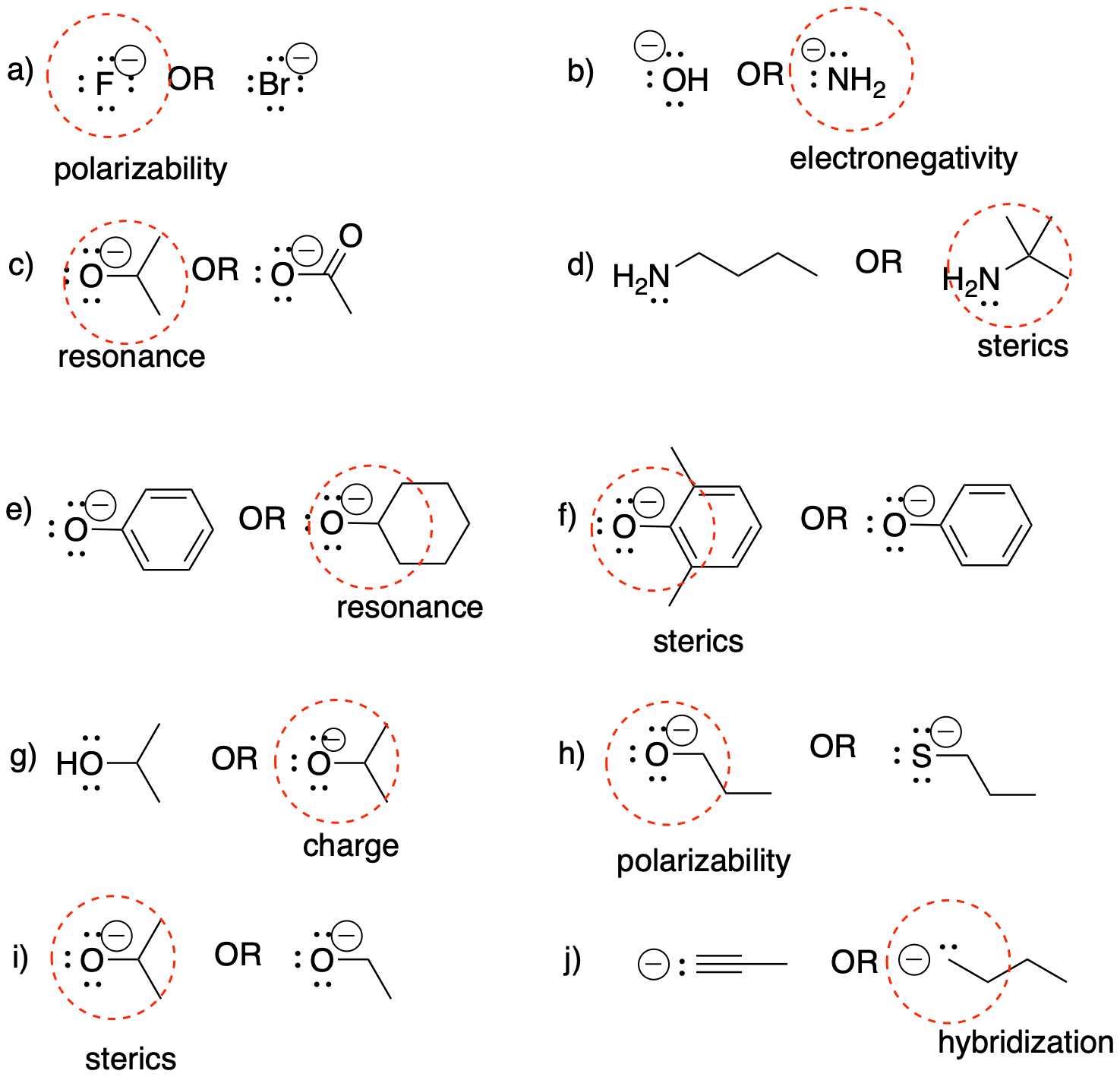
a) very strong b) strong c) weak (resonance) d) very strong e) weak (neutral)
f) weak (polarisable) g) weak (resonance) h) very strong i) weak (polarisable) j) weak (neutral)
k) medium-weak (C anion but sp) l) weak (neutral) m) weak (resonance) n) weak (polarisable) o) strong
p) weak (O anion but delocalised) q) weak (polarisable) r) very strong s) weak (polarisable) t) strong
Problem NS12.5.
An E1 mechanism can still occur with a dilute base such as 0.1 M NaOH. The reason has to do with relative concentrations and their influence on reaction ratses. If [HO-] is low, the rate of hydroxide colliding with the alkyl halide is very slow, and ionization may occur before the proton is removed. In these cases, the cation may initially be deprotonated by a lone pair on water because it is present at much higher concentration than the base. The resulting hydronium ion is then deprotonated by the base.
Problem NS12.6.
Additional anion-stabilizing factors can lower the basicity of a compound. There are two factors that stabilize the anion in cyanide, NC-, relative to methyllithium. The carbon in cyanide is sp hybridized compared to sp3 hybridized in methyllithium. Additional s character in an orbital lowers its energy, because at a given principal energy level the s orbitals are below the p orbitals in energy. Also, the electronegative nitrogen adds an inductive stabilizing effect for cyanide. These two stabilizing factors lower the basicity of cyanide to the level of a weak base.
Problem NS12.7.
Problem NS12.8.
Acetylides can undergo addition or elimination just like alkoxides, but their ability to undergo substitution is better than expected based on their basicity because of sterics. The anionic end of an acetylide has two linear carbons in a row. These linear carbons form a narrow point on the molecule, lowering the steric crowding that would certainly lead to elimination given the pKa of a terminal alkyne.
Problem NS12.9.
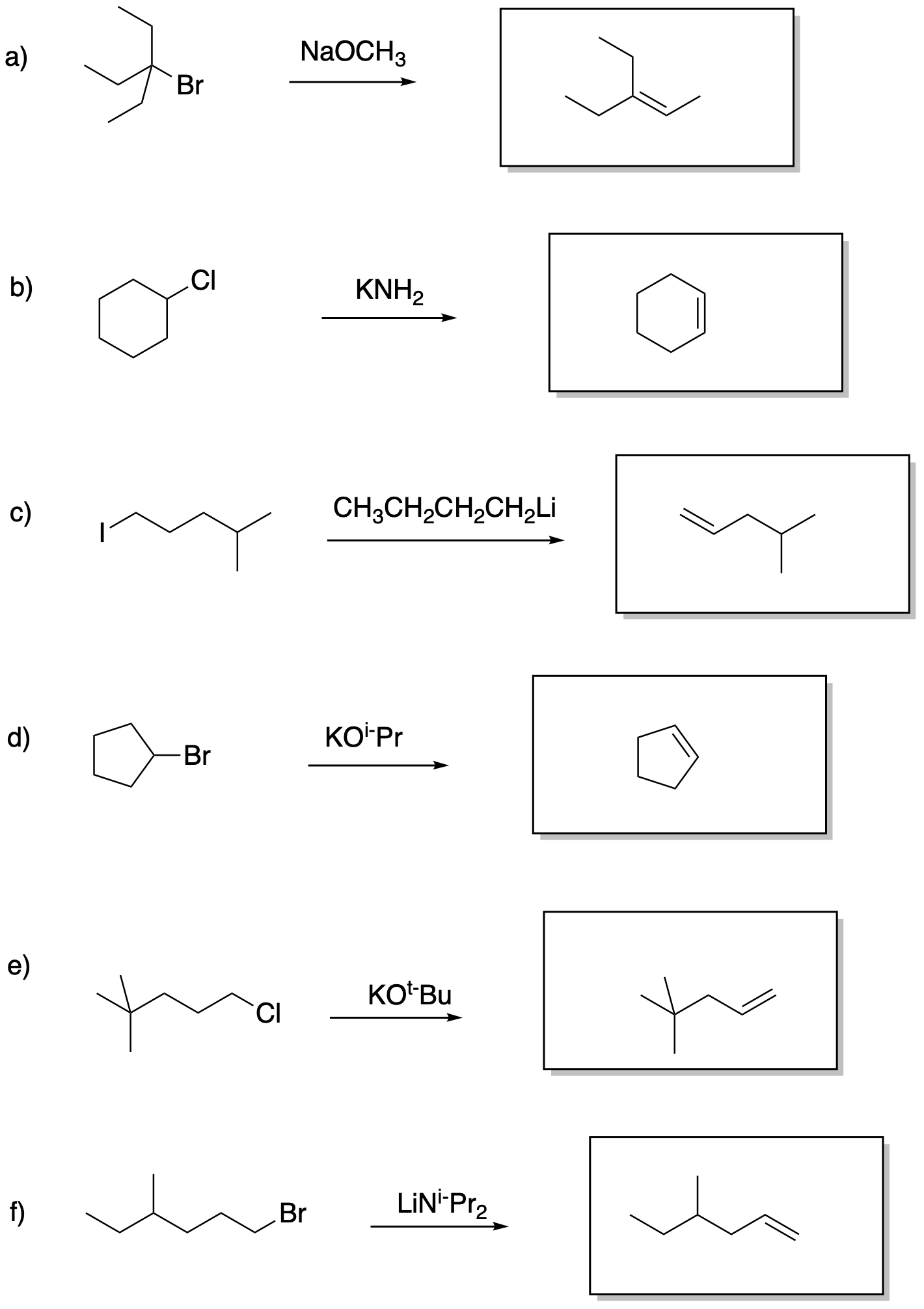
Problem NS13.1.
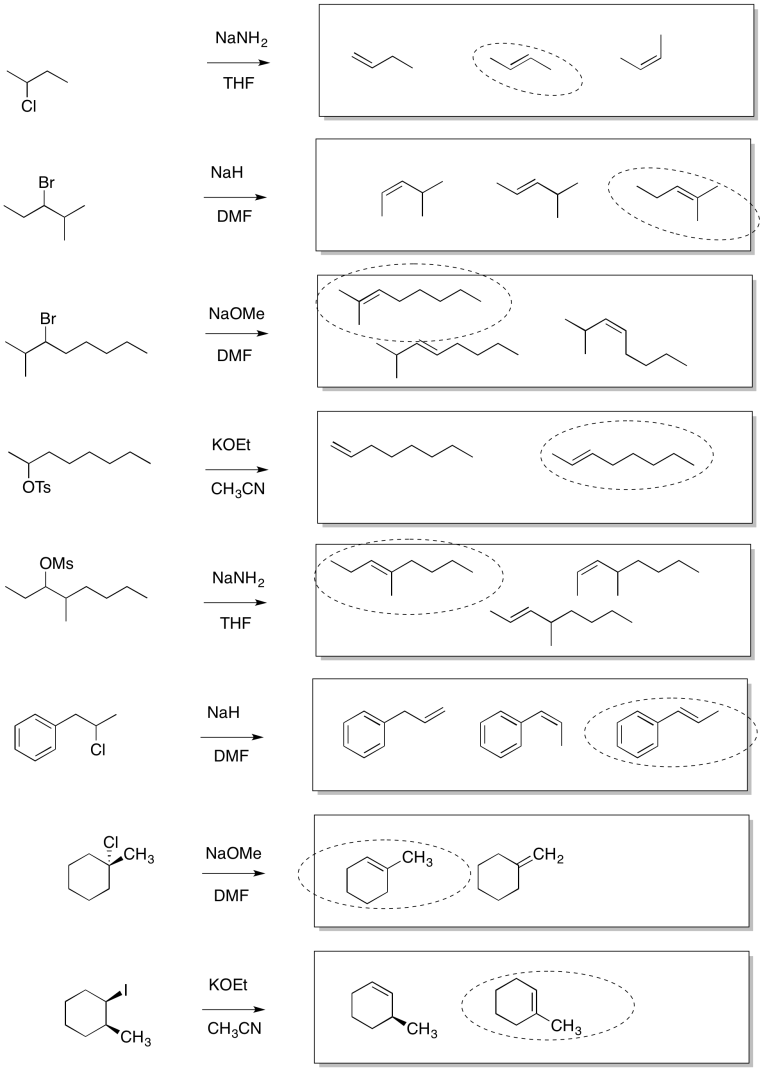
Problem NS13.2.
Problem NS14.1.
a) This is the (1S,2S) isomer.
b) This is the Z isomer because Br on one end of the double bond is on the same side as phenyl on the other end.
c) This appears to be the (1S,2R) isomer or the (1R,2S) isomer. Those two things are the same, so this is also described as the meso-isomer
d) This is the E isomer because Br on one end of the double bond is on the opposite side as phenyl on the other end.
Problem NS14.2.
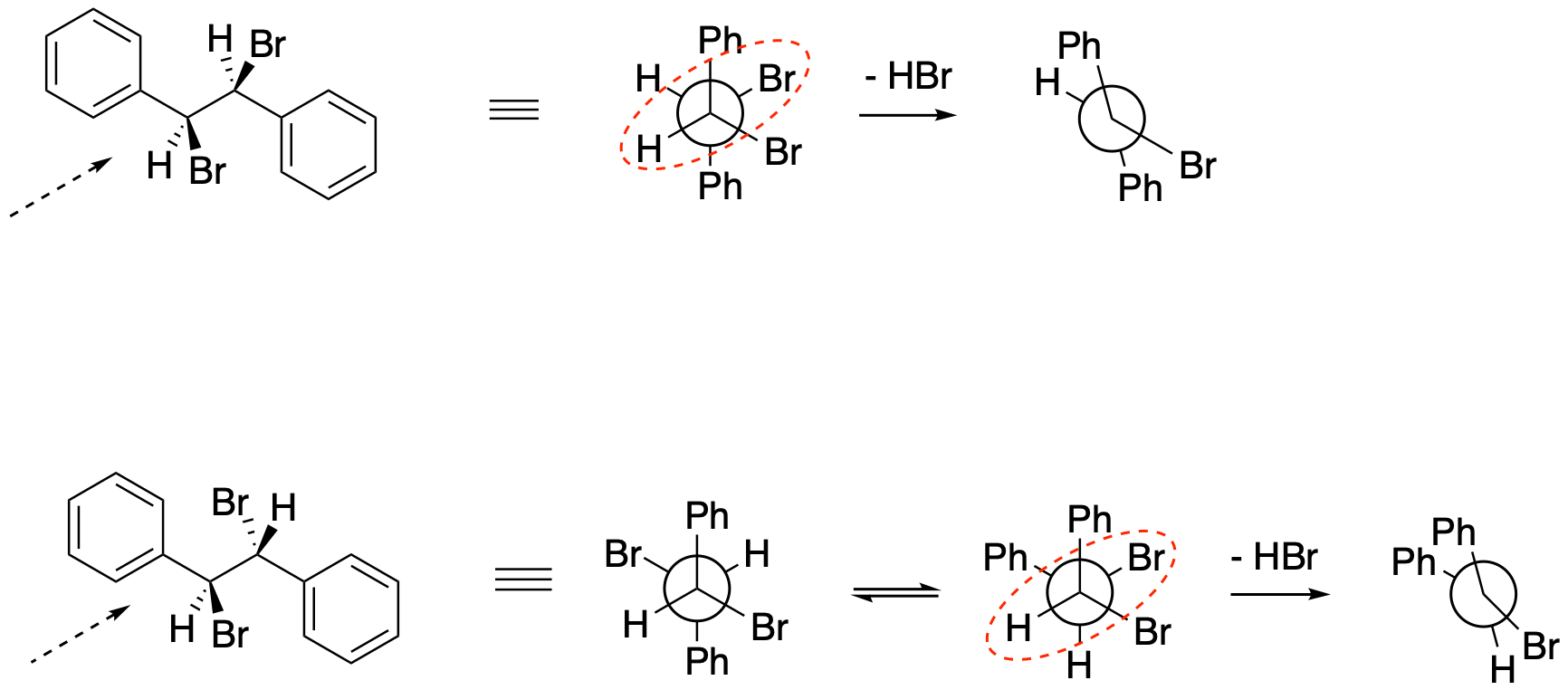
Problem NS14.3.
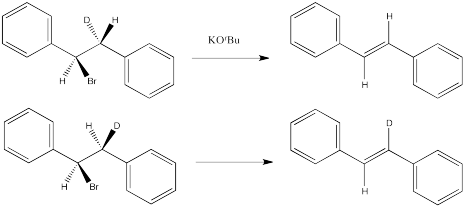
Problem NS14.4.
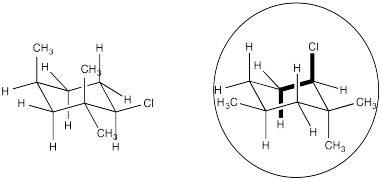
Problem NS14.5.
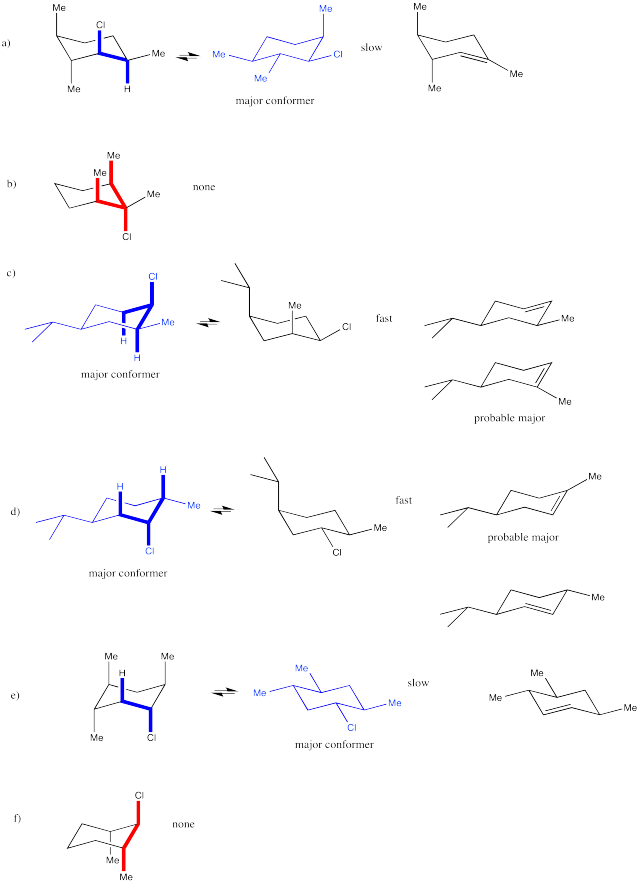
Problem NS14.6.
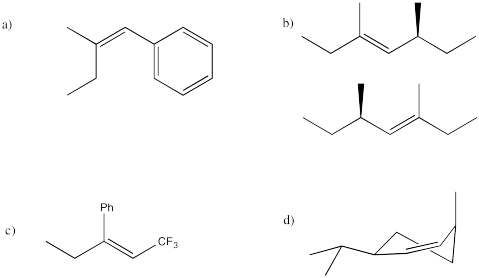
Problem NS14.7.
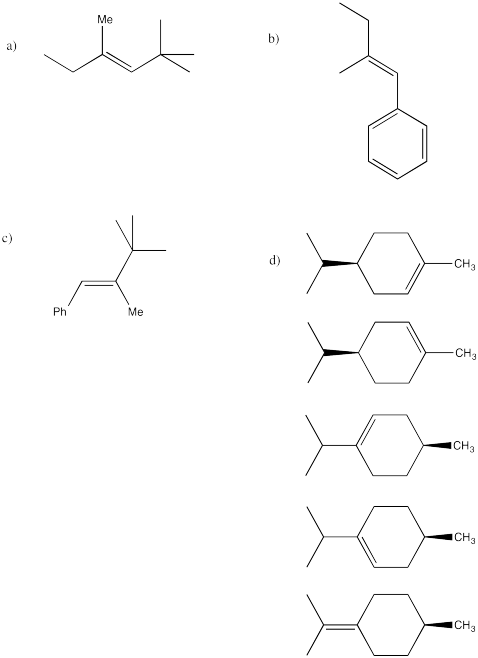
Problem 15.1.
Cation stability is important in an E1 reaction.
Problem 15.2.
Any tertiary alkyl halide would be a good example. Benzylic alkyl halides would also be good examples if they are either secondary or tertiary.
Problem 15.3.
Protic solvents could promote E1 reactions.
Problem 15.4.
A strong base could promote an E2 reaction.
Problem NS15.5.
Problem NS16.1.
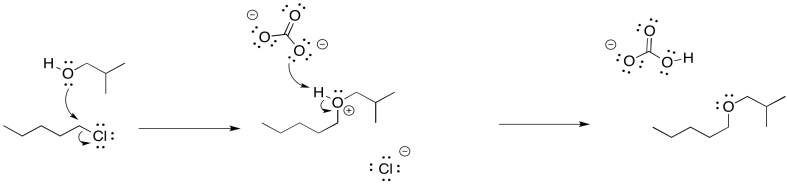
Problem NS16.2.
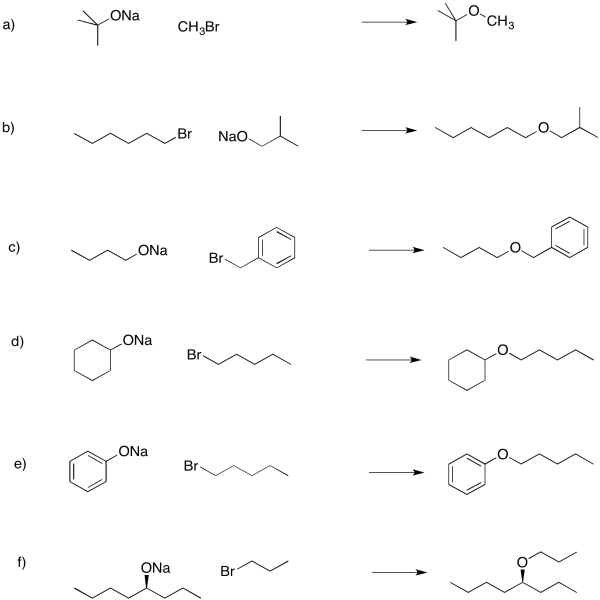
Problem NS16.3.
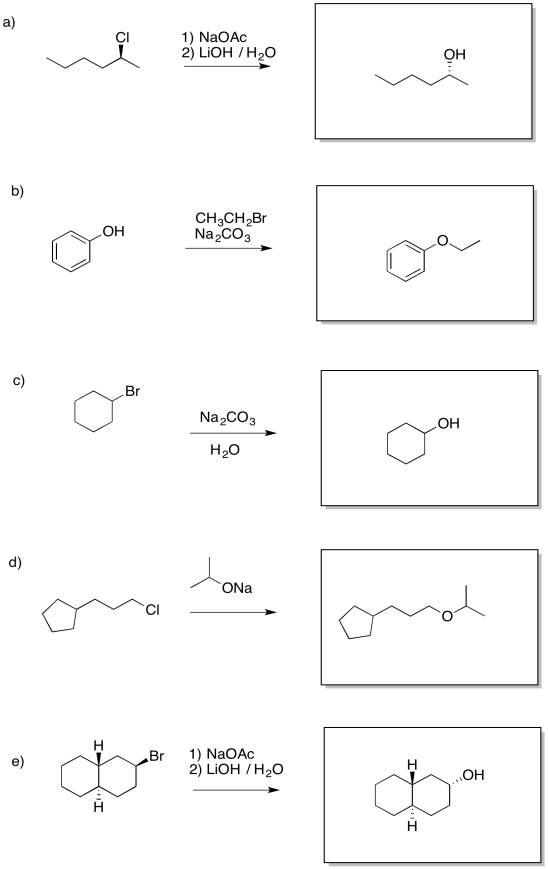
Problem NS17.1.
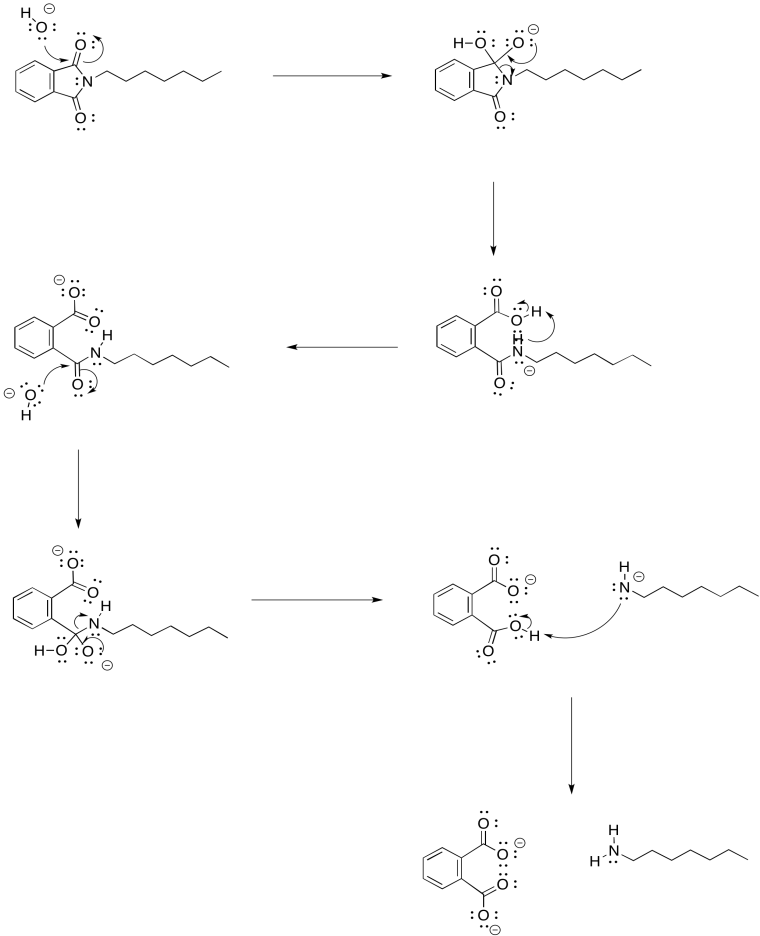
Problem NS17.2.
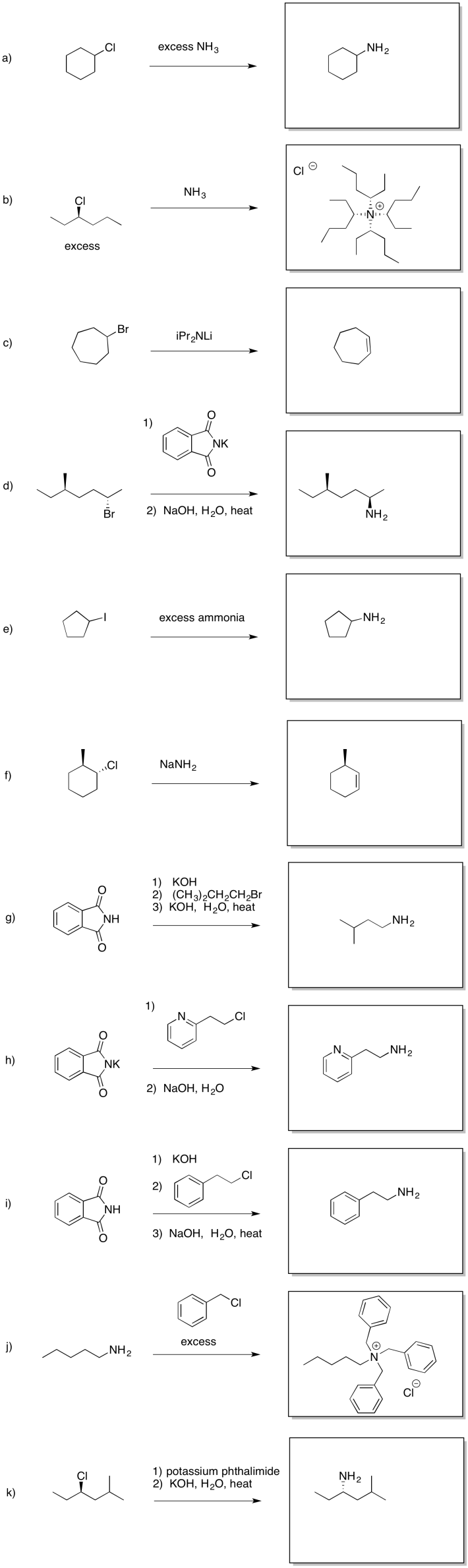
Problem NS17.3.
a) 1 HO- 2 HS- 3 H2O; the anions are more nucleophilic than the neutral, but sulfur is more polarizable and more stable than oxygen as an anion
b) 2 H2O 3 H3O+ 1 NH3; the neutrals are more nucleophilic than the cation, but nitrogen is less electronegative than oxygen
c) 1 CH3CH2NH2 2 (CH3)2CHNH2 3 (CH3)3CNH2; steric effects
Problem NS17.4.
a)
b)
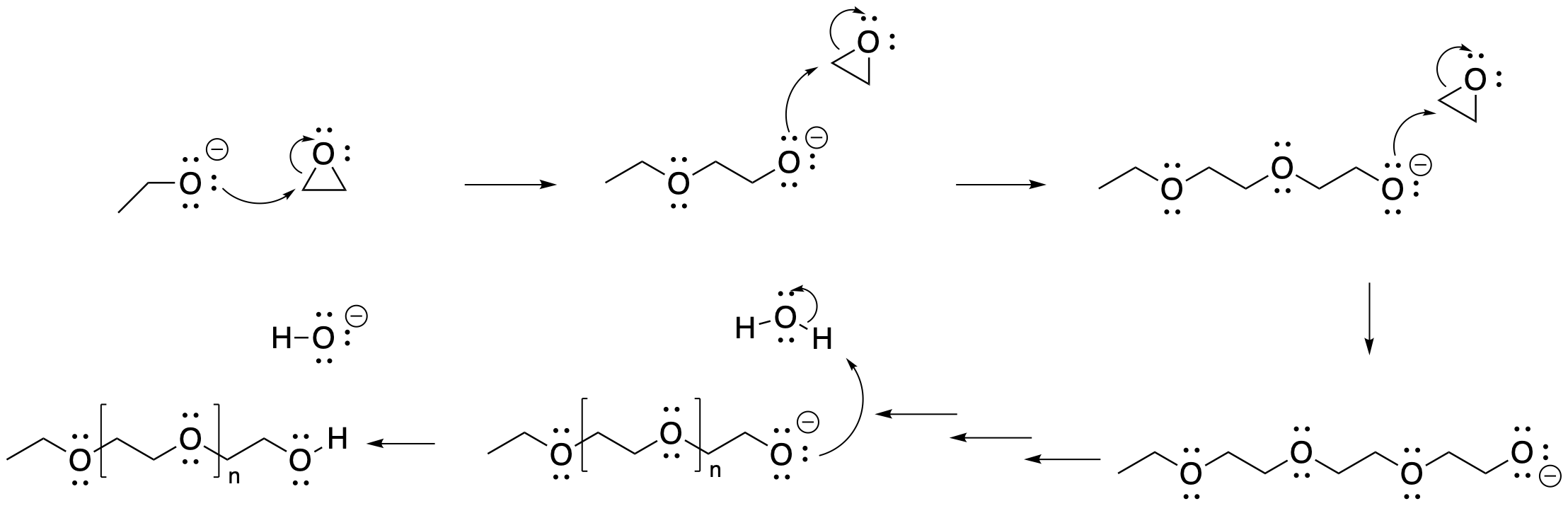
c) Ring strain promotes opening of the ring.
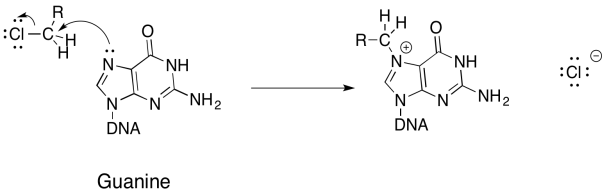
Problem NS18.1.
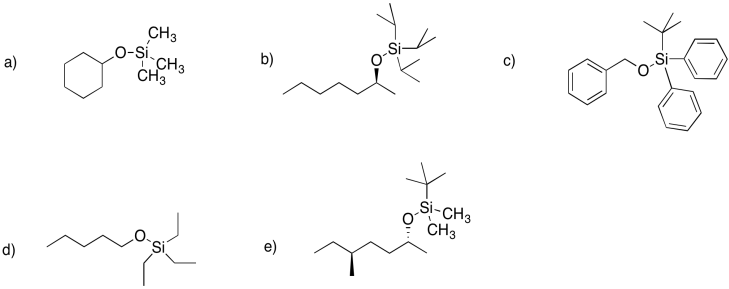
Problem NS18.2.
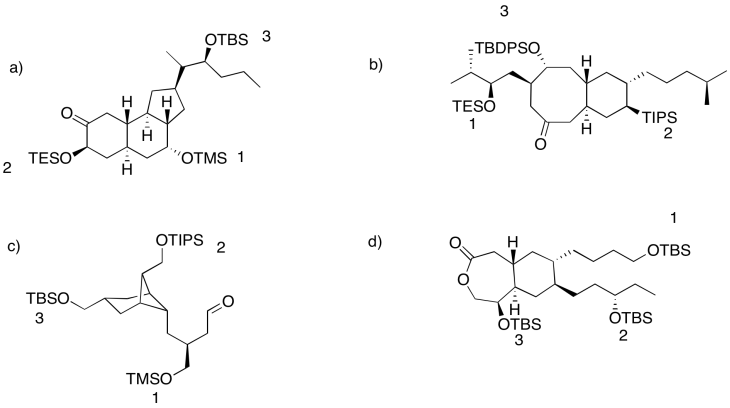
Problem NS18.3.
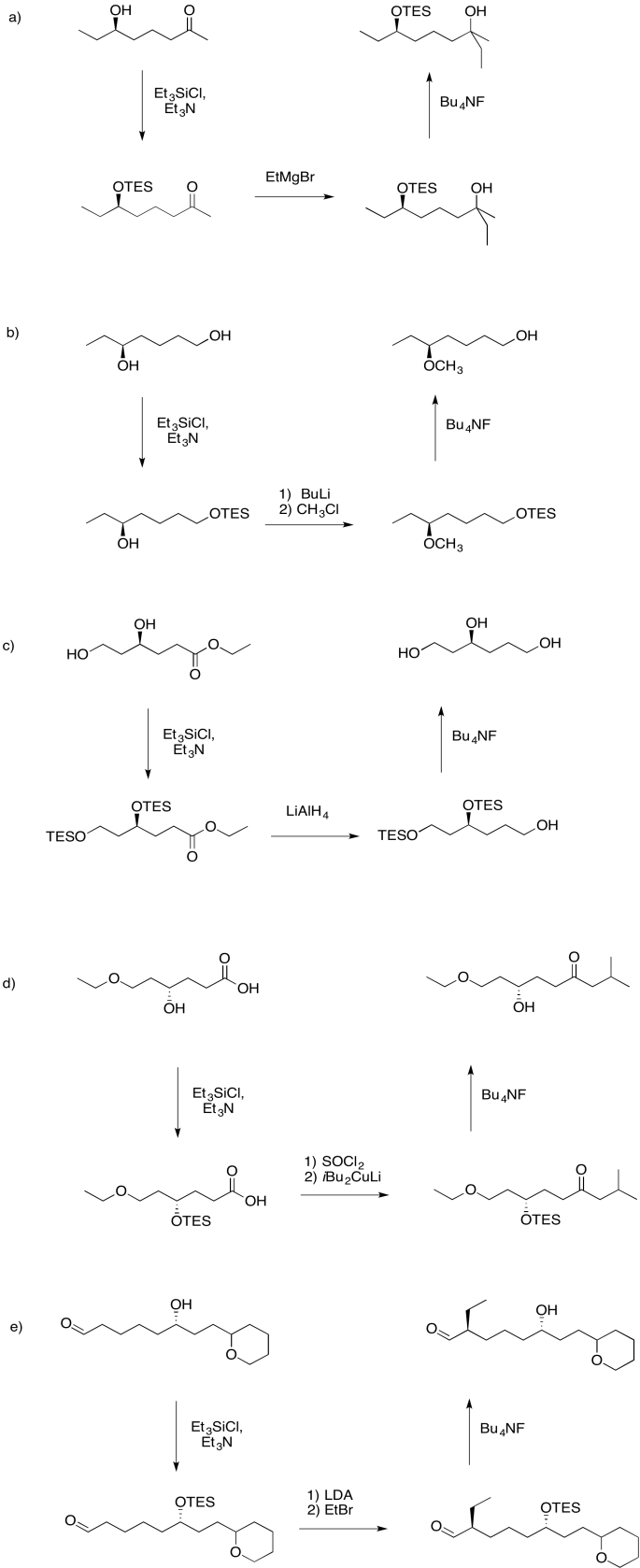
Problem NS19.1.
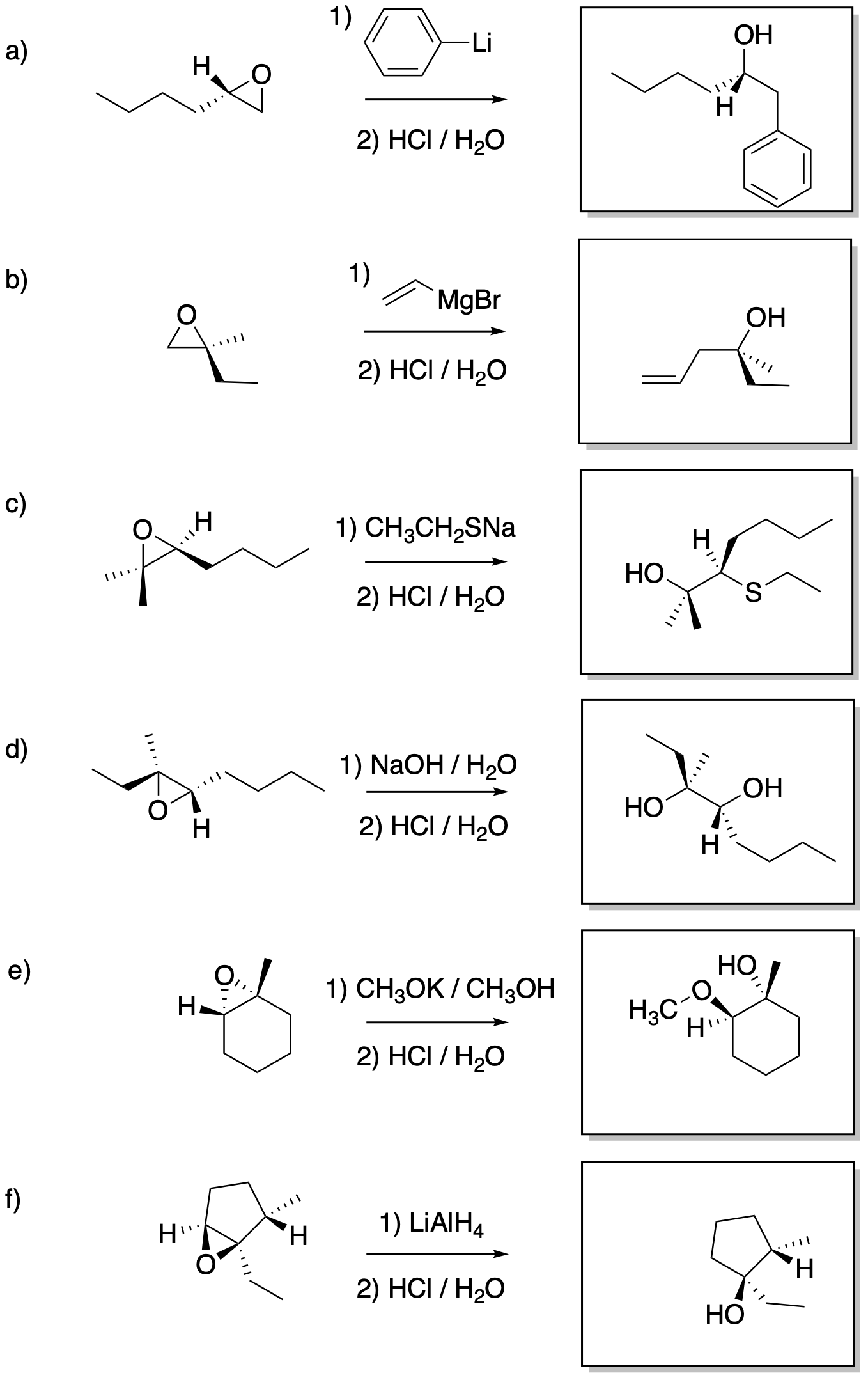
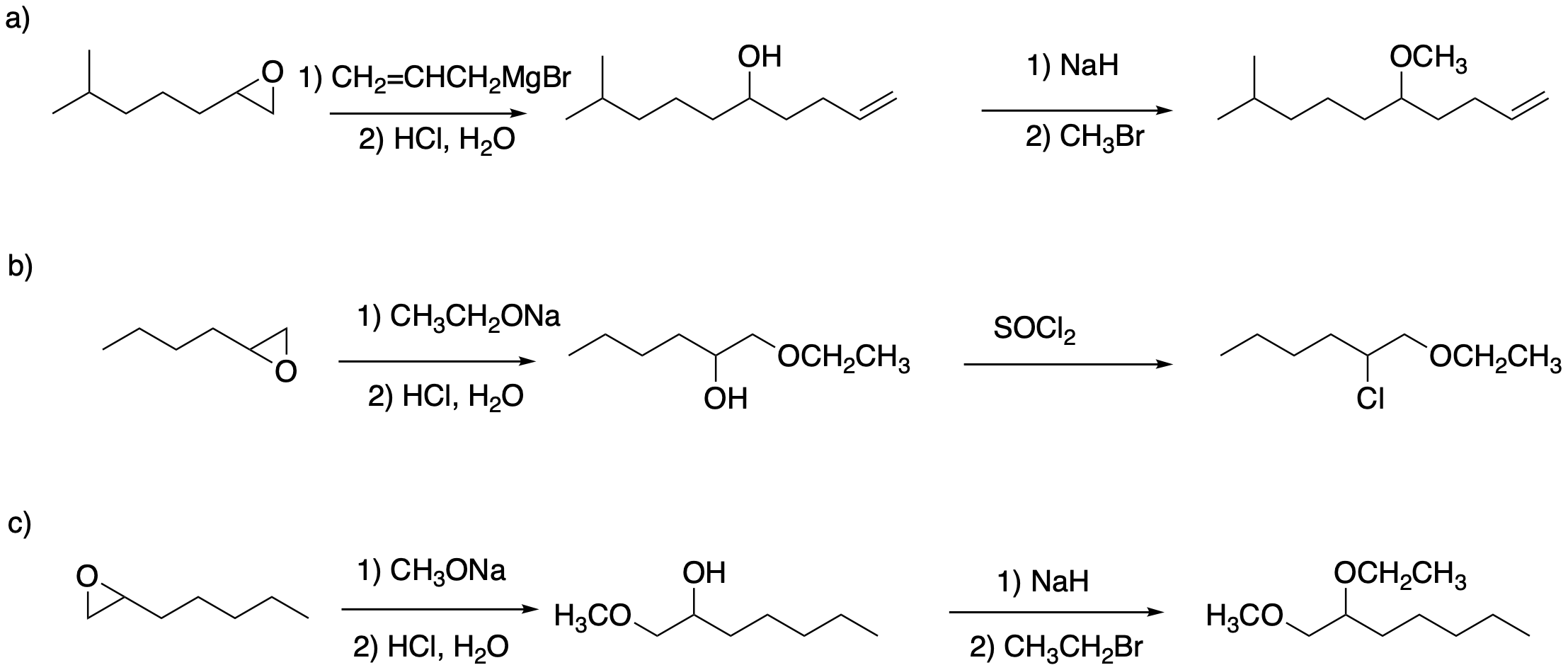
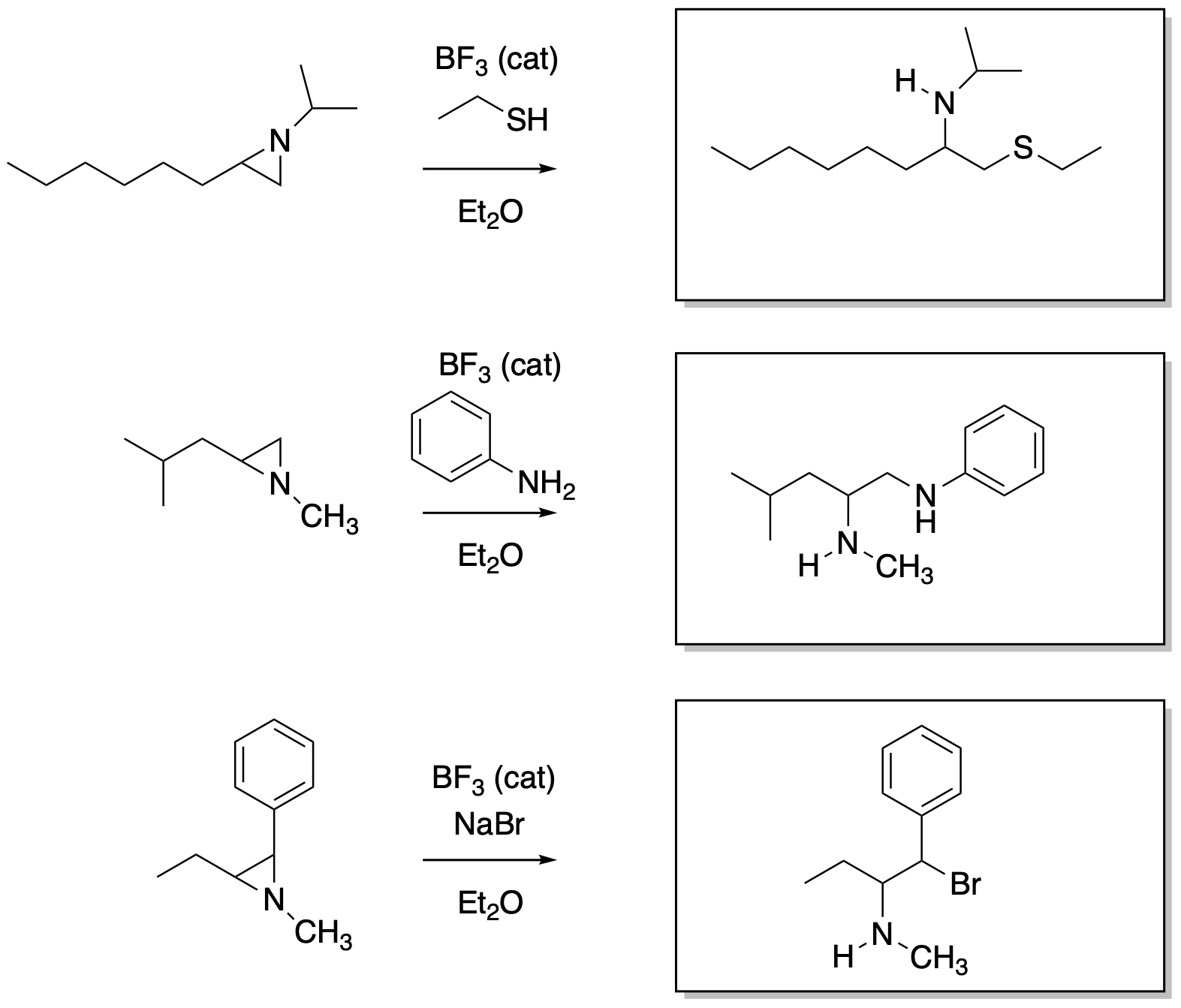
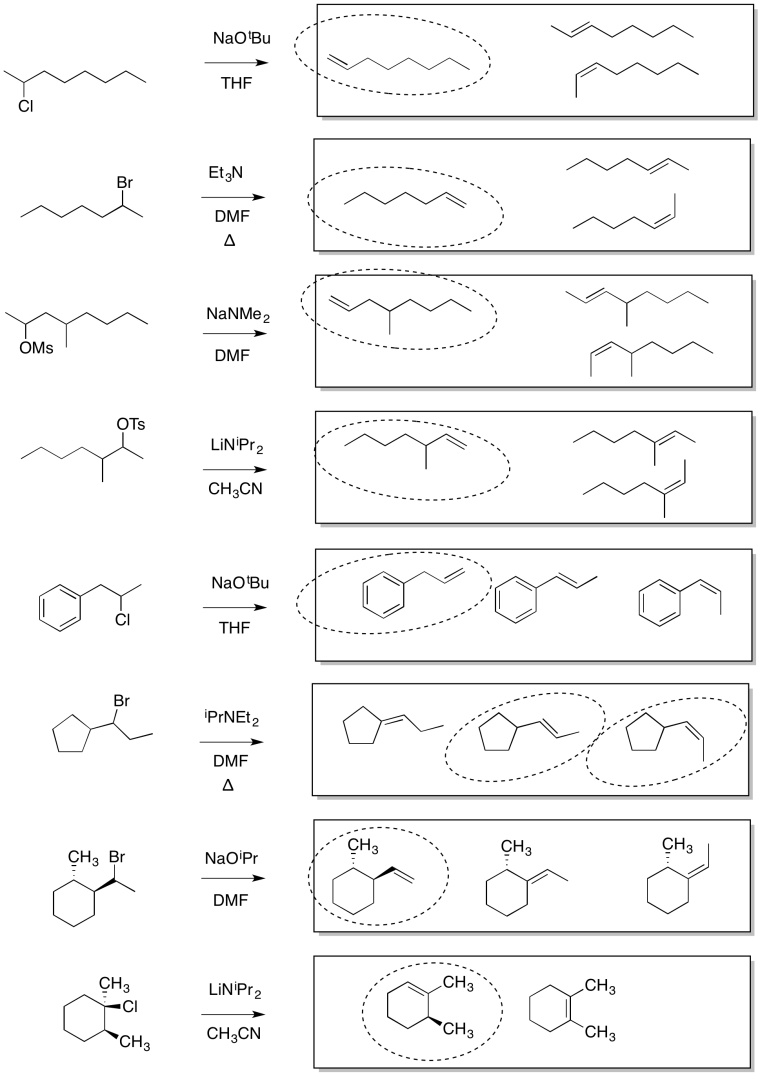
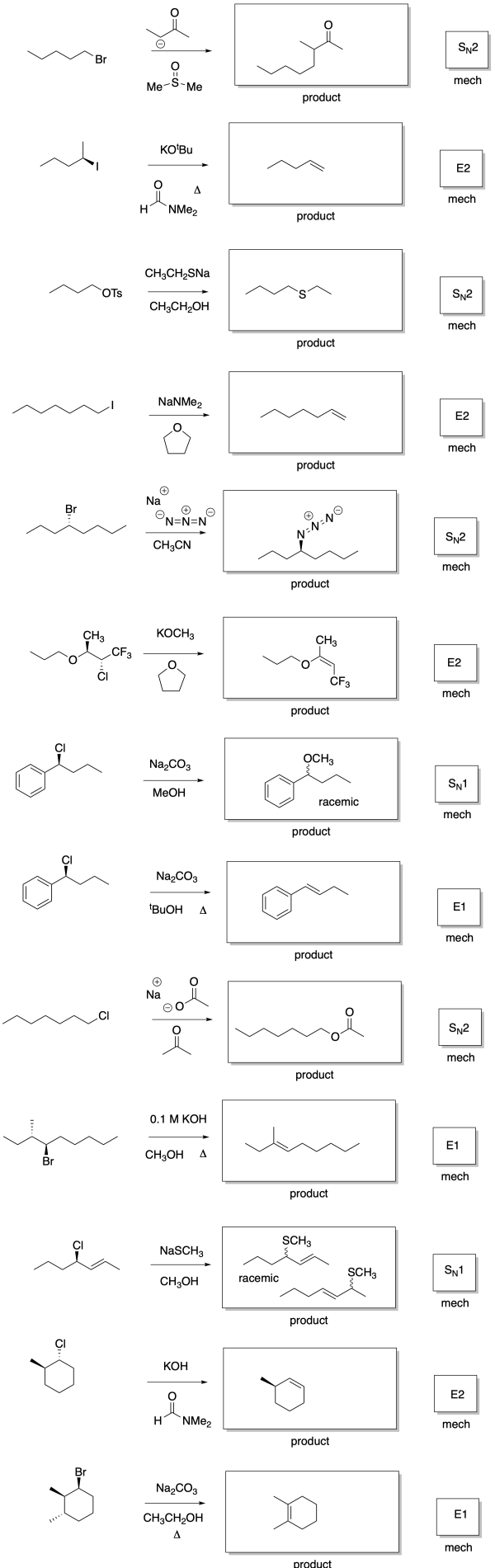
Answers may assume aqueous workup after the reagents shown. Only one answer shown per box; similar answers may also work.
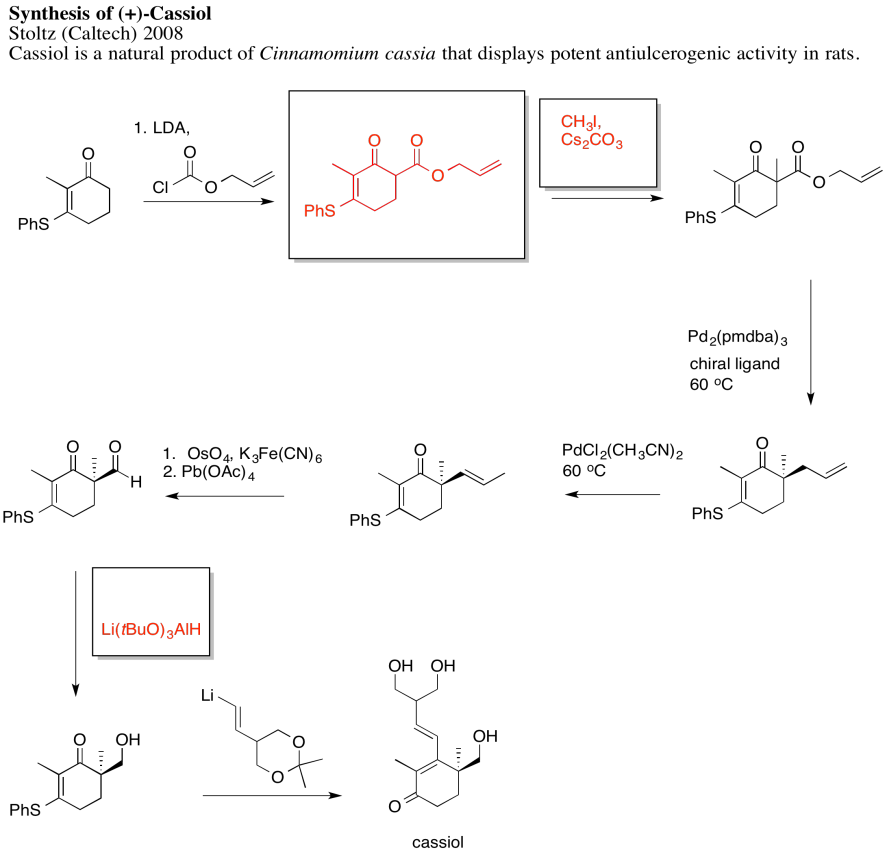
Problem NS19.2.
Answers may assume aqueous workup after the reagents shown. Only one answer shown per box; similar answers may also work.
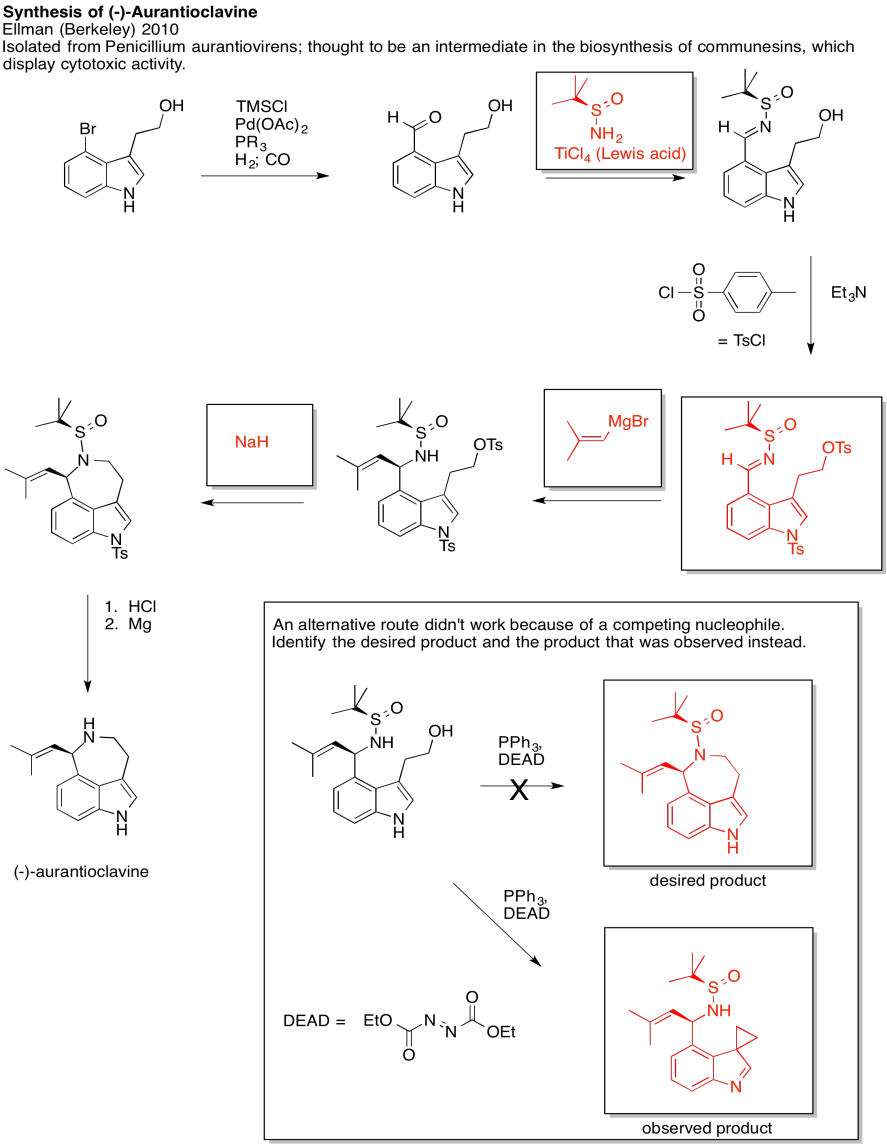
Problem NS19.3.
Answers may assume aqueous workup after the reagents shown. Only one answer shown per box; similar answers may also work.
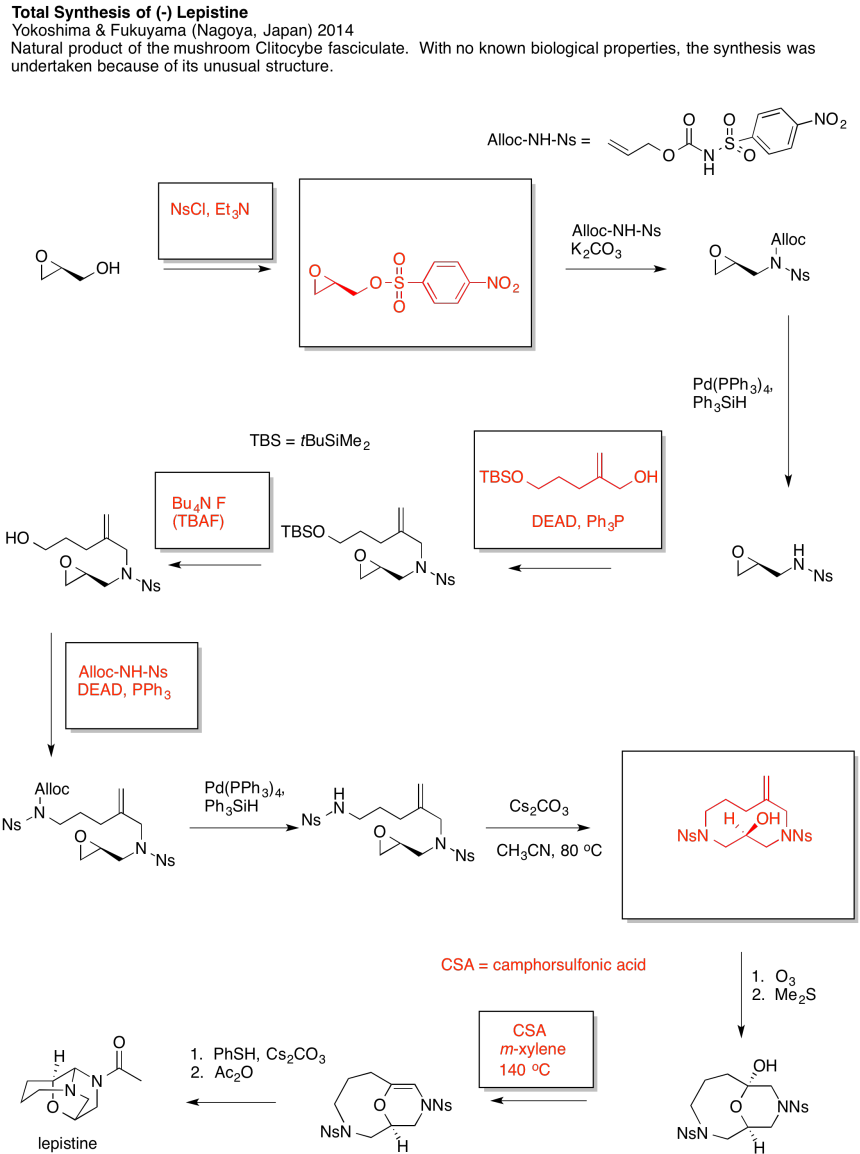
Problem NS19.4.
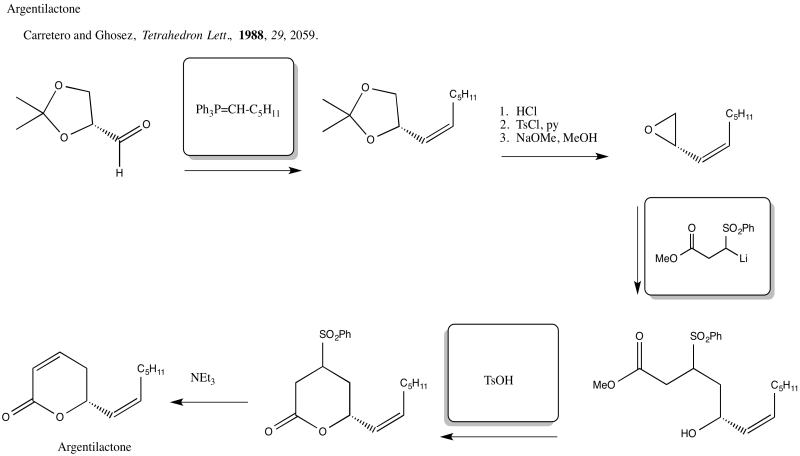
Problem NS19.5.
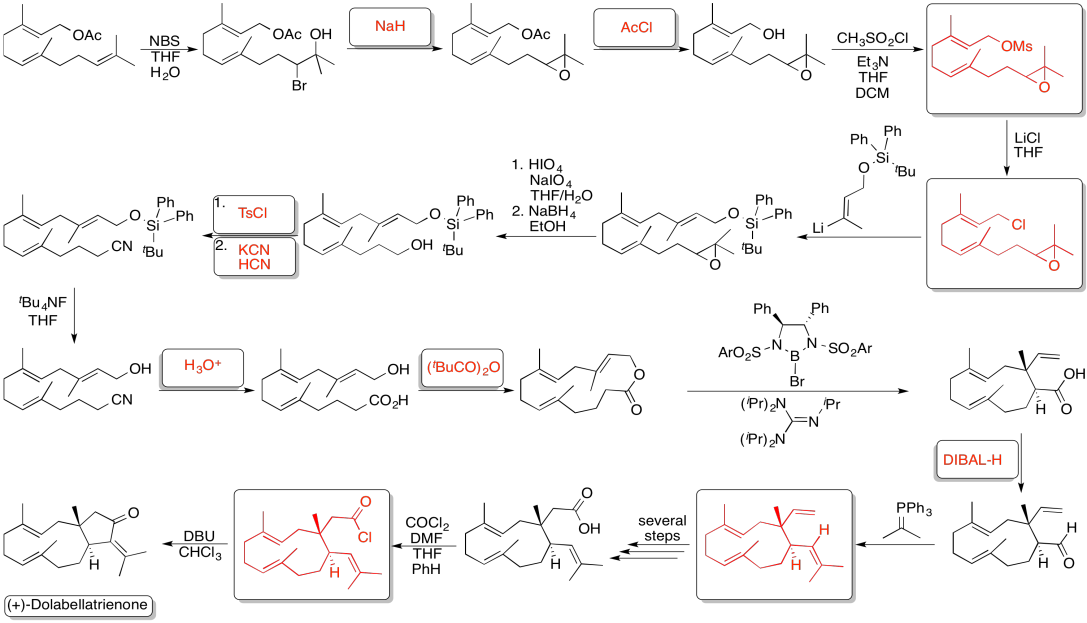
Problem NS19.6.
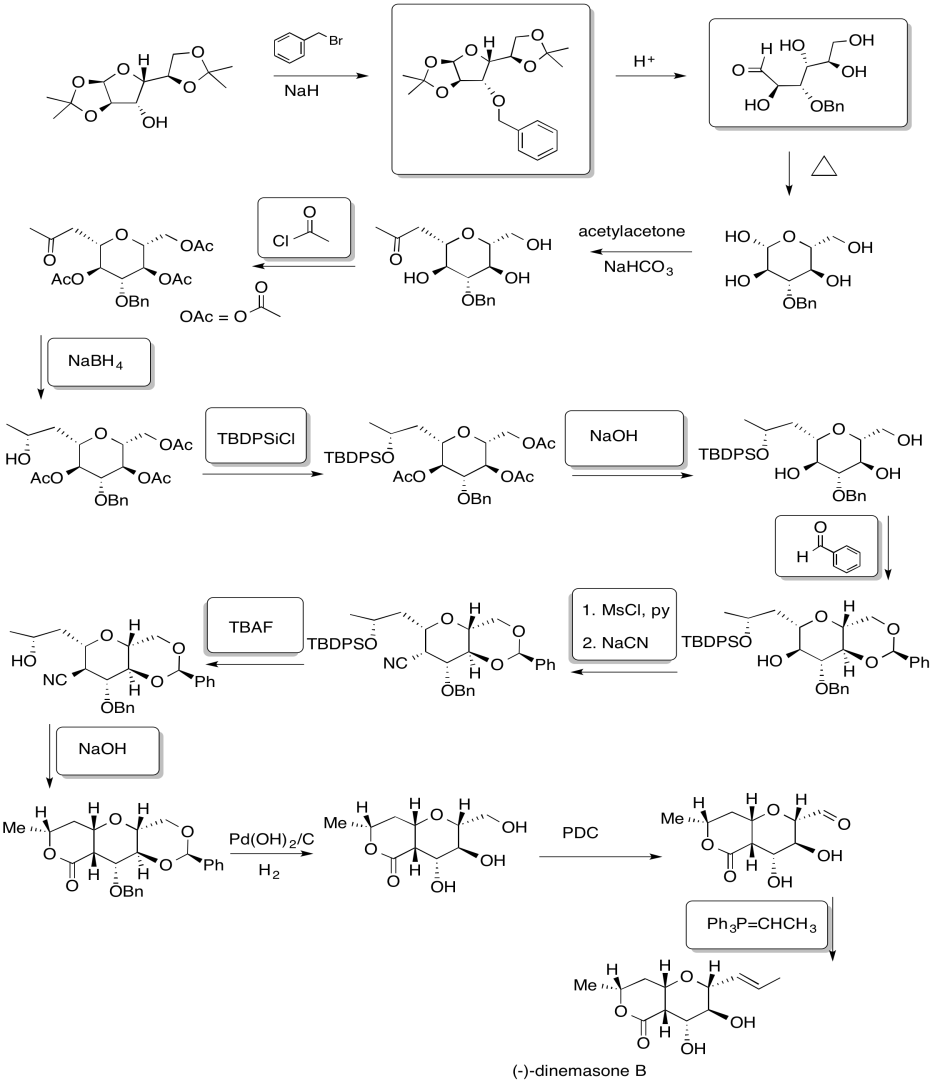
Problem NS19.7.
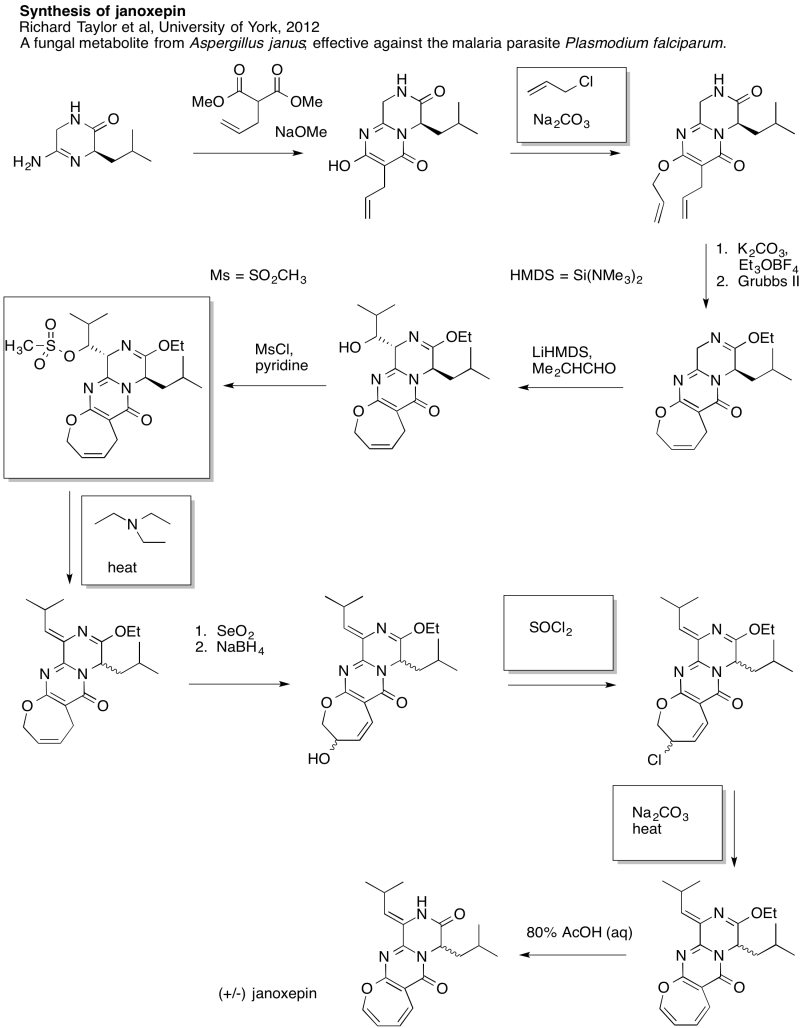
Problem NS19.8.
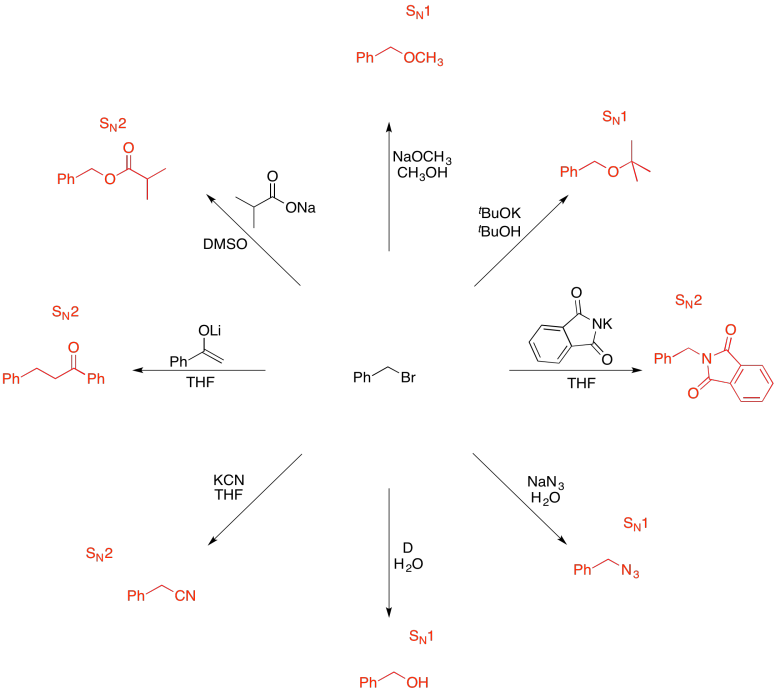
Problem NS19.9.
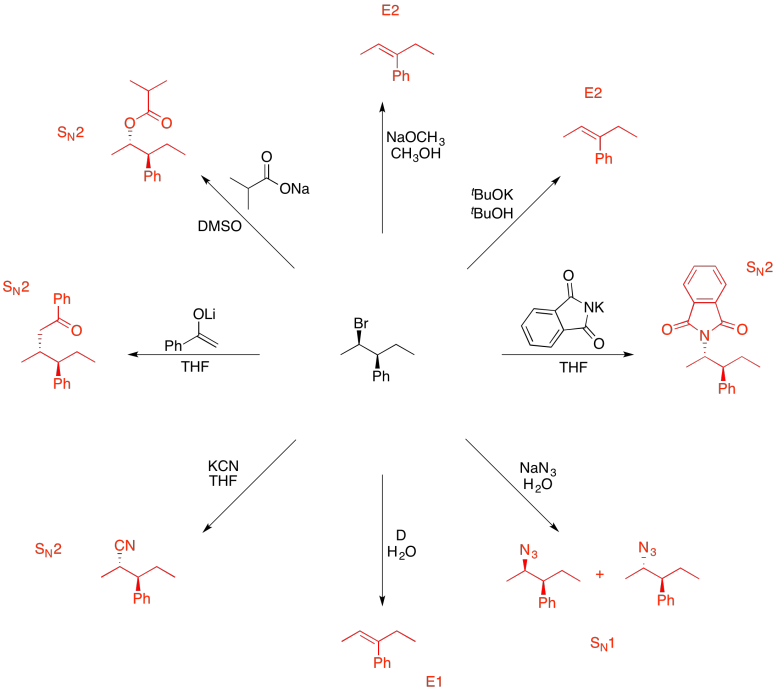
Problem NS19.10.
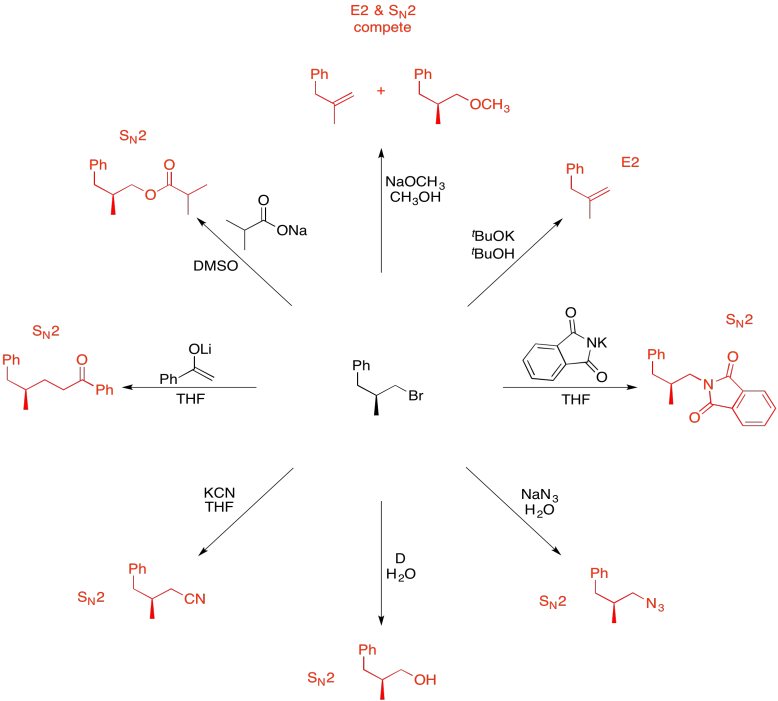
Problem NS19.11.
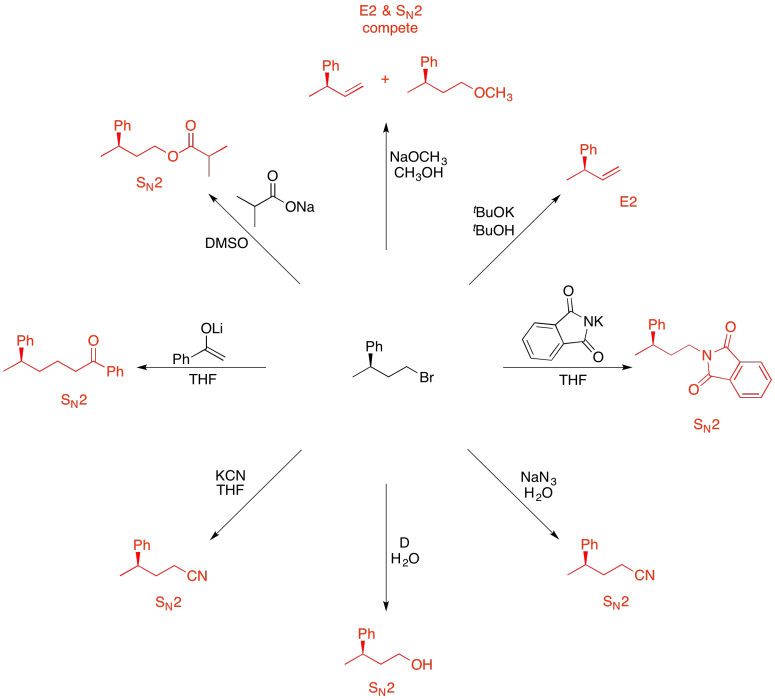
Problem NS19.12.
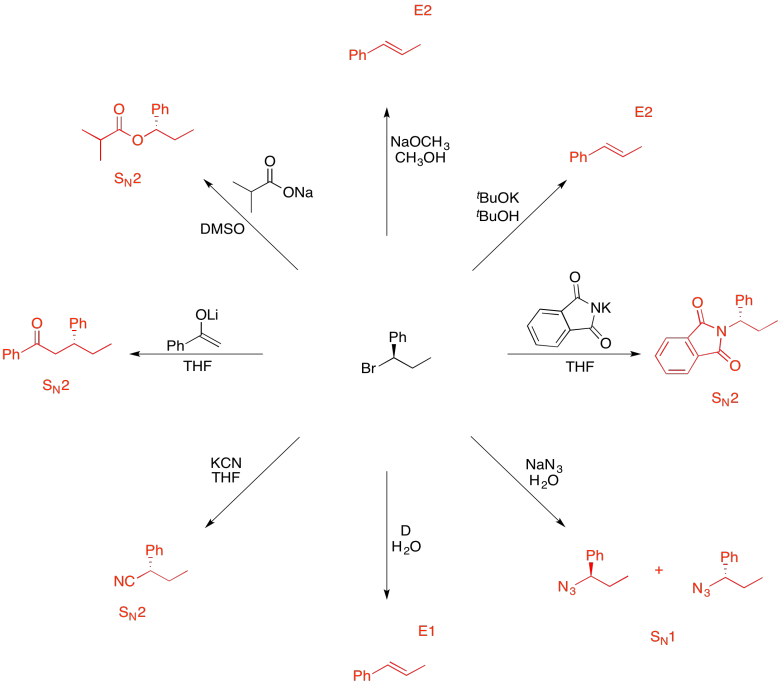
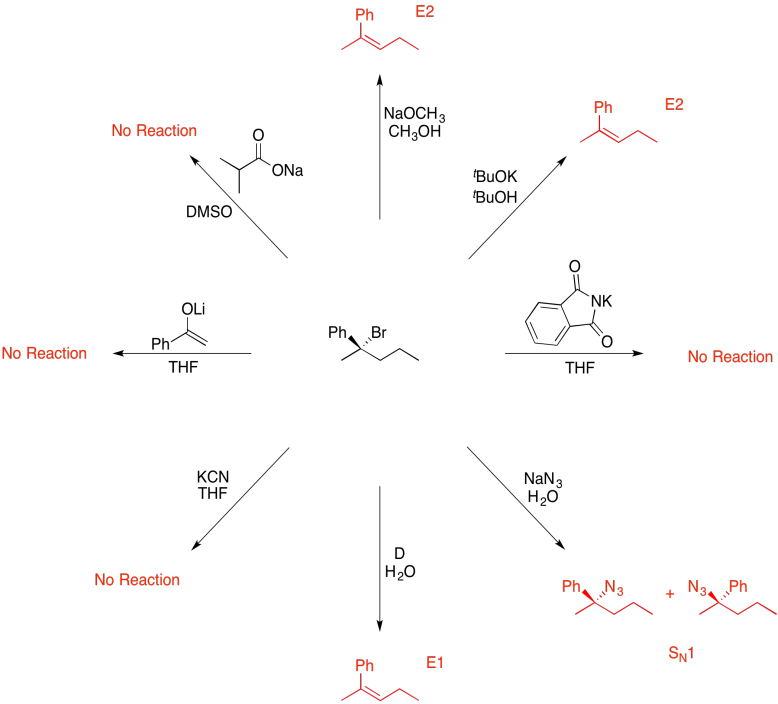
Problem NS19.13.
This site was written by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's University (retired) with other authors as noted). It is freely available for educational use.

Structure & Reactivity in Organic, Biological and Inorganic Chemistry by Chris Schaller is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported License.
Send corrections to cschaller@csbsju.edu
This material is based upon work supported by the National Science Foundation under Grant No. 1043566.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Navigation: